Issue published July 1, 2009 Previous issue | Next issue

- Volume 119, Issue 7
Go to section:
On the cover: Managing miscreant microbes

-
Science in Medicine
×
Hepatitis C virus versus innate and adaptive immune responses: a tale of coevolution and coexistence
Abstract
Since the identification of the hepatitis C virus (HCV) 20 years ago, much progress has been made in our understanding of its life cycle and interaction with the host immune system. Much has been learned from HCV itself, which, via decades of coevolution, gained an intricate knowledge of host innate and adaptive immune responses and developed sophisticated ways to preempt, subvert, and antagonize them. This review discusses the clinical, virological, and immunological features of acute and chronic hepatitis C and the role of the immune response in spontaneous and treatment-induced HCV clearance.
Authors
Barbara Rehermann


-
Review Series
×
Abstract
Intermediate filaments (IFs) are encoded by the largest gene family among the three major cytoskeletal protein groups. Unique IF compliments are expressed in selective cell types, and this expression is reflected in their involvement, upon mutation, as a cause of or predisposition to more than 80 human tissue-specific diseases. This Review Series covers diseases and functional and structural aspects pertaining to IFs and highlights the molecular and functional consequences of IF-associated diseases (IF-pathies). Exciting challenges and opportunities face the IF field, including developing both a better understanding of the pathogenesis of IF-pathies and targeted therapeutic approaches.
Authors
M. Bishr Omary
×Abstract
It took more than 100 years before it was established that the proteins that form intermediate filaments (IFs) comprise a unified protein family, the members of which are ubiquitous in virtually all differentiated cells and present both in the cytoplasm and in the nucleus. However, during the past 2 decades, knowledge regarding the functions of these structures has been expanding rapidly. Many disease-related roles of IFs have been revealed. In some cases, the molecular mechanisms underlying these diseases reflect disturbances in the functions traditionally assigned to IFs, i.e., maintenance of structural and mechanical integrity of cells and tissues. However, many disease conditions seem to link to the nonmechanical functions of IFs, many of which have been defined only in the past few years.
Authors
John E. Eriksson, Thomas Dechat, Boris Grin, Brian Helfand, Melissa Mendez, Hanna-Mari Pallari, Robert D. Goldman
×Abstract
Intermediate filaments (IFs) are major constituents of the cytoskeleton and nuclear boundary in animal cells. They are of prime importance for the functional organization of structural elements. Depending on the cell type, morphologically similar but biochemically distinct proteins form highly viscoelastic filament networks with multiple nanomechanical functions. Besides their primary role in cell plasticity and their established function as cellular stress absorbers, recently discovered gene defects have elucidated that structural alterations of IFs can affect their involvement both in signaling and in controlling gene regulatory networks. Here, we highlight the basic structural and functional properties of IFs and derive a concept of how mutations may affect cellular architecture and thereby tissue construction and physiology.
Authors
Harald Herrmann, Sergei V. Strelkov, Peter Burkhard, Ueli Aebi
×Abstract
Epidermolysis bullosa (EB) simplex is a rare genetic condition typified by superficial bullous lesions that result from frictional trauma to the skin. Most cases are due to dominantly acting mutations in either keratin 14 (K14) or K5, the type I and II intermediate filament (IF) proteins tasked with forming a pancytoplasmic network of 10-nm filaments in basal keratinocytes of the epidermis and in other stratified epithelia. Defects in K5/K14 filament network architecture cause basal keratinocytes to become fragile and account for their trauma-induced rupture. Here we review how laboratory investigations centered on keratin biology have deepened our understanding of the etiology and pathophysiology of EB simplex and revealed novel avenues for its therapy.
Authors
Pierre A. Coulombe, Michelle L. Kerns, Elaine Fuchs
×Abstract
Simple epithelial keratins (SEKs) are found primarily in single-layered simple epithelia and include keratin 7 (K7), K8, K18–K20, and K23. Genetically engineered mice that lack SEKs or overexpress mutant SEKs have helped illuminate several keratin functions and served as important disease models. Insight into the contribution of SEKs to human disease has indicated that K8 and K18 are the major constituents of Mallory-Denk bodies, hepatic inclusions associated with several liver diseases, and are essential for inclusion formation. Furthermore, mutations in the genes encoding K8, K18, and K19 predispose individuals to a variety of liver diseases. Hence, as we discuss here, the SEK cytoskeleton is involved in the orchestration of several important cellular functions and contributes to the pathogenesis of human liver disease.
Authors
M. Bishr Omary, Nam-On Ku, Pavel Strnad, Shinichiro Hanada
×Abstract
Muscle fiber deterioration resulting in progressive skeletal muscle weakness, heart failure, and respiratory distress occurs in more than 20 inherited myopathies. As discussed in this Review, one of the newly identified myopathies is desminopathy, a disease caused by dysfunctional mutations in desmin, a type III intermediate filament protein, or αB-crystallin, a chaperone for desmin. The range of clinical manifestations in patients with desminopathy is wide and may overlap with those observed in individuals with other myopathies. Awareness of this disease needs to be heightened, diagnostic criteria reliably outlined, and molecular testing readily available; this would ensure prevention of sudden death from cardiac arrhythmias and other complications.
Authors
Lev G. Goldfarb, Marinos C. Dalakas
×Abstract
Intermediate filaments (IFs) are abundant structures found in most eukaryotic cells, including those in the nervous system. In the CNS, the primary components of neuronal IFs are α-internexin and the neurofilament triplet proteins. In the peripheral nervous system, a fifth neuronal IF protein known as peripherin is also present. IFs in astrocytes are primarily composed of glial fibrillary acidic protein (GFAP), although vimentin is also expressed in immature astrocytes and some mature astrocytes. In this Review, we focus on the IFs of glial cells (primarily GFAP) and neurons as well as their relationship to different neurodegenerative diseases.
Authors
Ronald K.H. Liem, Albee Messing
×Abstract
The main function of the nuclear lamina, an intermediate filament meshwork lying primarily beneath the inner nuclear membrane, is to provide structural scaffolding for the cell nucleus. However, the lamina also serves other functions, such as having a role in chromatin organization, connecting the nucleus to the cytoplasm, gene transcription, and mitosis. In somatic cells, the main protein constituents of the nuclear lamina are lamins A, C, B1, and B2. Interest in the nuclear lamins increased dramatically in recent years with the realization that mutations in LMNA, the gene encoding lamins A and C, cause a panoply of human diseases (“laminopathies”), including muscular dystrophy, cardiomyopathy, partial lipodystrophy, and progeroid syndromes. Here, we review the laminopathies and the long strange trip from basic cell biology to therapeutic approaches for these diseases.
Authors
Howard J. Worman, Loren G. Fong, Antoine Muchir, Stephen G. Young
×Abstract
Intermediate filaments (IFs) are a key component of the cytoskeleton in virtually all vertebrate cells, including those of the lens of the eye. IFs help integrate individual cells into their respective tissues. This Review focuses on the lens-specific IF proteins beaded filament structural proteins 1 and 2 (BFSP1 and BFSP2) and their role in lens physiology and disease. Evidence generated in studies in both mice and humans suggests a critical role for these proteins and their filamentous polymers in establishing the optical properties of the eye lens and in maintaining its transparency. For instance, mutations in both BFSP1 and BFSP2 cause cataract in humans. We also explore the potential role of BFSP1 and BFSP2 in aging processes in the lens.
Authors
Shuhua Song, Andrew Landsbury, Ralf Dahm, Yizhi Liu, Qingjiong Zhang, Roy A. Quinlan





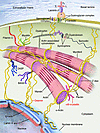



-
Commentaries
×
Abstract
Immature spermatozoa are vulnerable to oxidative stress after their release from the testes, due in part to an innate deficiency in antioxidant enzymes. The male reproductive tract compensates for this deficiency by secreting antioxidant enzymes such as glutathione peroxidase 5 (Gpx5) into the epididymal lumen. In this issue of the JCI, Chabory et al. examined the phenotype of Gpx5–/– mice and found that while deletion of this gene did not seem to affect fertility per se, it did influence the incidence of miscarriage and embryonic defects in mated wild-type female mice (see the related article beginning on page 2074). Importantly, the appearance of these problems was age dependent and associated with signs of oxidative stress in the spermatozoa. These results demonstrate the key importance of Gpx5 as an extracellular antioxidant designed to protect maturing mammalian spermatozoa from oxidative stress.
Authors
R. John Aitken
×Abstract
Current therapies for immune-mediated diseases, such as rheumatoid arthritis and MS, could represent the proverbial bird in the hand — a known entity, yet limited in potential. Emerging biologic therapeutics for these diseases carry with them the potential for known as well as unknown adverse effects. Alemtuzumab, a biologic that depletes leukocytes, shows great promise for the treatment of MS. However, a significant number of patients develop autoimmunity after treatment, raising the level of caution for the use of this drug. In this issue of the JCI, Jones et al. describe a link between IL-21 levels and alemtuzumab-associated autoimmunity (see the related article beginning on page 2052). They show that proliferation of lymphocytes in those patients with autoimmunity is higher than in those without autoimmunity and suggest that the lymphopenia-driven proliferation of T cells, in combination with higher IL-21 levels, results in autoimmunity. This study helps inspire new enthusiasm for making a grab for the proverbial two birds in the bush — representing undiscovered therapies — with greater confidence.
Authors
Terri M. Laufer, Gregory F. Wu
×Abstract
The paradoxical ability to launch effective immunity against pathogens while avoiding the horror autotoxicus of autoimmunity is one of the most remarkable features of the mammalian immune system. This assumes a particular evolutionary significance at the maternal/fetal interface, where avoidance of immune reactivity to the fetus is vital to the propagation of the species itself. Several mechanisms of suppressing maternal immunity against the fetus have been described. The article by Collins et al. in this issue of the JCI describes a novel mechanism of avoiding immune surveillance in which the migratory capacity of dendritic cells at the maternal/fetal interface is restrained (see the related article beginning on page 2062).
Authors
Rana Chakraborty, Bali Pulendran


-
Research Articles
×
Abstract
Hepatic fibrosis develops as a response to chronic liver injury and almost exclusively occurs in a proinflammatory environment. However, the role of inflammatory mediators in fibrogenic responses of the liver is only poorly understood. We therefore investigated the role of CC chemokines and their receptors in hepatic fibrogenesis. The CC chemokines MIP-1α, MIP-1β, and RANTES and their receptors CCR1 and CCR5 were strongly upregulated in 2 experimental mouse models of fibrogenesis. Neutralization of CC chemokines by the broad-spectrum CC chemokine inhibitor 35k efficiently reduced hepatic fibrosis, and CCR1- and CCR5-deficient mice displayed substantially reduced hepatic fibrosis and macrophage infiltration. Analysis of fibrogenesis in CCR1- and CCR5-chimeric mice revealed that CCR1 mediates its profibrogenic effects in BM-derived cells, whereas CCR5 mediates its profibrogenic effects in resident liver cells. CCR5 promoted hepatic stellate cell (HSC) migration through a redox-sensitive, PI3K-dependent pathway. Both CCR5-deficient HSCs and CCR1- and CCR5-deficient Kupffer cells displayed strong suppression of CC chemokine–induced migration. Finally, we detected marked upregulation of RANTES, CCR1, and CCR5 in patients with hepatic cirrhosis, confirming activation of the CC chemokine system in human fibrogenesis. Our data therefore support a role for the CC chemokine system in hepatic fibrogenesis and suggest distinct roles for CCR1 and CCR5 in Kupffer cells and HSCs.
Authors
Ekihiro Seki, Samuele De Minicis, Geum-Youn Gwak, Johannes Kluwe, Sayaka Inokuchi, Christina A. Bursill, Josep M. Llovet, David A. Brenner, Robert F. Schwabe
×Abstract
Maintenance of vascular integrity is critical for homeostasis, and temporally and spatially regulated vascular leak is a central feature of inflammation. Sphingosine-1-phosphate (S1P) can regulate endothelial barrier function, but the sources of the S1P that provide this activity in vivo and its importance in modulating different inflammatory responses are unknown. We report here that mutant mice engineered to selectively lack S1P in plasma displayed increased vascular leak and impaired survival after anaphylaxis, administration of platelet-activating factor (PAF) or histamine, and exposure to related inflammatory challenges. Increased leak was associated with increased interendothelial cell gaps in venules and was reversed by transfusion with wild-type erythrocytes (which restored plasma S1P levels) and by acute treatment with an agonist for the S1P receptor 1 (S1pr1). S1pr1 agonist did not protect wild-type mice from PAF-induced leak, consistent with plasma S1P levels being sufficient for S1pr1 activation in wild-type mice. However, an agonist for another endothelial cell Gi-coupled receptor, Par2, did protect wild-type mice from PAF-induced vascular leak, and systemic treatment with pertussis toxin prevented rescue by Par2 agonist and sensitized wild-type mice to leak-inducing stimuli in a manner that resembled the loss of plasma S1P. Our results suggest that the blood communicates with blood vessels via plasma S1P to maintain vascular integrity and regulate vascular leak. This pathway prevents lethal responses to leak-inducing mediators in mouse models.
Authors
Eric Camerer, Jean B. Regard, Ivo Cornelissen, Yoga Srinivasan, Daniel N. Duong, Daniel Palmer, Trung H. Pham, Jinny S. Wong, Rajita Pappu, Shaun R. Coughlin
×Abstract
The formation of polyploid cells is part of the developmental program of several tissues. During postnatal development, binucleated tetraploid cells arise in the liver, caused by failure in cytokinesis. In this report, we have shown that the initiation of cytokinesis failure events and the subsequent appearance of binucleated tetraploid cells are strictly controlled by the suckling-to-weaning transition in rodents. We found that daily light/dark rhythms and carbohydrate intake did not affect liver tetraploidy. In contrast, impairment of insulin signaling drastically reduced the formation of binucleated tetraploid cells, whereas repeated insulin injections promoted the generation of these liver cells. Furthermore, inhibition of Akt activity decreased the number of cytokinesis failure events, possibly through the mammalian target of rapamycin signaling complex 2 (mTORC2), which indicates that the PI3K/Akt pathway lies downstream of the insulin signal to regulate the tetraploidization process. To our knowledge, these results are the first demonstration in a physiological context that insulin signaling through Akt controls a specific cell division program and leads to the physiologic generation of binucleated tetraploid liver cells.
Authors
Séverine Celton-Morizur, Grégory Merlen, Dominique Couton, Germain Margall-Ducos, Chantal Desdouets
×Abstract
Heterozygous mutations in the gene encoding the pancreatic homeodomain transcription factor pancreatic duodenal homeobox 1 (PDX1) are associated with maturity onset diabetes of the young, type 4 (MODY4) and type 2 diabetes. Pdx1 governs the early embryonic development of the pancreas and the later differentiation of the insulin-producing islet β cells of the endocrine compartment. We derived a Pdx1 hypomorphic allele that reveals a role for Pdx1 in the specification of endocrine progenitors. Mice homozygous for this allele displayed a selective reduction in endocrine lineages associated with decreased numbers of endocrine progenitors and a marked reduction in levels of mRNA encoding the proendocrine transcription factor neurogenin 3 (Ngn3). During development, Pdx1 occupies an evolutionarily conserved enhancer region of Ngn3 and interacts with the transcription factor one cut homeobox 1 (Hnf6) to activate this enhancer. Furthermore, mRNA levels of all 4 members of the transcription factor network that regulates Ngn3 expression, SRY-box containing gene 9 (Sox9), Hnf6, Hnf1b, and forkhead box A2 (Foxa2), were decreased in homozygous mice. Pdx1 also occupied regulatory sequences in Foxa2 and Hnf1b. Thus, Pdx1 contributes to specification of endocrine progenitors both by regulating expression of Ngn3 directly and by participating in a cross-regulatory transcription factor network during early pancreas development. These results provide insights that may be applicable to β cell replacement strategies involving the guided differentiation of ES cells or other progenitor cell types into the β cell lineage, and they suggest a molecular mechanism whereby human PDX1 mutations cause diabetes.
Authors
Jennifer M. Oliver-Krasinski, Margaret T. Kasner, Juxiang Yang, Michael F. Crutchlow, Anil K. Rustgi, Klaus H. Kaestner, Doris A. Stoffers
×Abstract
Microbial colonization of mucosal surfaces may be an initial event in the progression to disease, and it is often a transient process. For the extracellular pathogen Streptococcus pneumoniae studied in a mouse model, nasopharyngeal carriage is eliminated over a period of weeks and requires cellular rather than humoral immunity. Here, we demonstrate that primary infection led to TLR2-dependent recruitment of monocyte/macrophages into the upper airway lumen, where they engulfed pneumococci. Pharmacologic depletion of luminal monocyte/macrophages by intranasal instillation of liposomal clodronate diminished pneumococcal clearance. Efficient clearance of colonization required TLR2 signaling to generate a population of pneumococcal-specific IL-17–expressing CD4+ T cells. Depletion of either IL-17A or CD4+ T cells was sufficient to block the recruitment of monocyte/macrophages that allowed for effective late pneumococcal clearance. In contrast with naive mice, previously colonized mice showed enhanced early clearance that correlated with a more robust influx of luminal neutrophils. As for primary colonization, these cellular responses required Th17 immunity. Our findings demonstrate that monocyte/macrophages and neutrophils recruited to the mucosal surface are key effectors in clearing primary and secondary bacterial colonization, respectively.
Authors
Zhe Zhang, Thomas B. Clarke, Jeffrey N. Weiser
×Abstract
Influenza-related complications continue to be a major cause of mortality worldwide. Due to unclear mechanisms, a substantial number of influenza-related deaths result from bacterial superinfections, particularly secondary pneumococcal pneumonia. Here, we report what we believe to be a novel mechanism by which influenza-induced type I IFNs sensitize hosts to secondary bacterial infections. Influenza-infected mice deficient for type I IFN-α/β receptor signaling (Ifnar–/– mice) had improved survival and clearance of secondary Streptococcus pneumoniae infection from the lungs and blood, as compared with similarly infected wild-type animals. The less effective response in wild-type mice seemed to be attributable to impaired production of neutrophil chemoattractants KC (also known as Cxcl1) and Mip2 (also known as Cxcl2) following secondary challenge with S. pneumoniae. This resulted in inadequate neutrophil responses during the early phase of host defense against secondary bacterial infection. Indeed, influenza-infected wild-type mice cleared secondary pneumococcal pneumonia after pulmonary administration of exogenous KC and Mip2, whereas neutralization of Cxcr2, the common receptor for KC and Mip2, reversed the protective phenotype observed in Ifnar–/– mice. These data may underscore the importance of the type I IFN inhibitory pathway on CXC chemokine production. Collectively, these findings highlight what we believe to be a novel mechanism by which the antiviral response to influenza sensitizes hosts to secondary bacterial pneumonia.
Authors
Arash Shahangian, Edward K. Chow, Xiaoli Tian, Jason R. Kang, Amir Ghaffari, Su Y. Liu, John A. Belperio, Genhong Cheng, Jane C. Deng
×Abstract
Recognition of LPS by TLR4 on immune sentinel cells such as macrophages is thought to be key to the recruitment of neutrophils to sites of infection with Gram-negative bacteria. To explore whether endothelial TLR4 plays a role in this process, we engineered and imaged mice that expressed TLR4 exclusively on endothelium (known herein as EndotheliumTLR4 mice). Local administration of LPS into tissue induced comparable neutrophil recruitment in EndotheliumTLR4 and wild-type mice. Following systemic LPS or intraperitoneal E. coli administration, most neutrophils were sequestered in the lungs of wild-type mice and did not accumulate at primary sites of infection. In contrast, EndotheliumTLR4 mice showed reduced pulmonary capillary neutrophil sequestration over the first 24 hours; as a result, they mobilized neutrophils to primary sites of infection, cleared bacteria, and resisted a dose of E. coli that killed 50% of wild-type mice in the first 48 hours. In fact, the only defect we detected in EndotheliumTLR4 mice was a failure to accumulate neutrophils in the lungs following intratracheal administration of LPS; this response required TLR4 on bone marrow–derived immune cells. Therefore, endothelial TLR4 functions as the primary intravascular sentinel system for detection of bacteria, whereas bone marrow–derived immune cells are critical for pathogen detection at barrier sites. Nonendothelial TLR4 contributes to failure to accumulate neutrophils at primary infection sites in a disseminated systemic infection.
Authors
Graciela Andonegui, Hong Zhou, Daniel Bullard, Margaret M. Kelly, Sarah C. Mullaly, Braedon McDonald, Elizabeth M. Long, Stephen M. Robbins, Paul Kubes
×Abstract
The activation of type I IFN signaling is a major component of host defense against viral infection, but it is not typically associated with immune responses to extracellular bacterial pathogens. Using mouse and human airway epithelial cells, we have demonstrated that Staphylococcus aureus activates type I IFN signaling, which contributes to its virulence as a respiratory pathogen. This response was dependent on the expression of protein A and, more specifically, the Xr domain, a short sequence–repeat region encoded by DNA that consists of repeated 24-bp sequences that are the basis of an internationally used epidemiological typing scheme. Protein A was endocytosed by airway epithelial cells and subsequently induced IFN-β expression, JAK-STAT signaling, and IL-6 production. Mice lacking IFN-α/β receptor 1 (IFNAR-deficient mice), which are incapable of responding to type I IFNs, were substantially protected against lethal S. aureus pneumonia compared with wild-type control mice. The profound immunological consequences of IFN-β signaling, particularly in the lung, may help to explain the conservation of multiple copies of the Xr domain of protein A in S. aureus strains and the importance of protein A as a virulence factor in the pathogenesis of staphylococcal pneumonia.
Authors
Francis J. Martin, Marisa I. Gomez, Dawn M. Wetzel, Guido Memmi, Maghnus O’Seaghdha, Grace Soong, Christian Schindler, Alice Prince
×Abstract
Atrial fibrillation (AF), the most common human cardiac arrhythmia, is associated with abnormal intracellular Ca2+ handling. Diastolic Ca2+ release from the sarcoplasmic reticulum via “leaky” ryanodine receptors (RyR2s) is hypothesized to contribute to arrhythmogenesis in AF, but the molecular mechanisms are incompletely understood. Here, we have shown that mice with a genetic gain-of-function defect in Ryr2 (which we termed Ryr2R176Q/+ mice) did not exhibit spontaneous AF but that rapid atrial pacing unmasked an increased vulnerability to AF in these mice compared with wild-type mice. Rapid atrial pacing resulted in increased Ca2+/calmodulin-dependent protein kinase II (CaMKII) phosphorylation of RyR2, while both pharmacologic and genetic inhibition of CaMKII prevented AF inducibility in Ryr2R176Q/+ mice. This result suggests that AF requires both an arrhythmogenic substrate (e.g., RyR2 mutation) and enhanced CaMKII activity. Increased CaMKII phosphorylation of RyR2 was observed in atrial biopsies from mice with atrial enlargement and spontaneous AF, goats with lone AF, and patients with chronic AF. Genetic inhibition of CaMKII phosphorylation of RyR2 in Ryr2S2814A knockin mice reduced AF inducibility in a vagotonic AF model. Together, these findings suggest that increased RyR2-dependent Ca2+ leakage due to enhanced CaMKII activity is an important downstream effect of CaMKII in individuals susceptible to AF induction.
Authors
Mihail G. Chelu, Satyam Sarma, Subeena Sood, Sufen Wang, Ralph J. van Oort, Darlene G. Skapura, Na Li, Marco Santonastasi, Frank Ulrich Müller, Wilhelm Schmitz, Ulrich Schotten, Mark E. Anderson, Miguel Valderrábano, Dobromir Dobrev, Xander H.T. Wehrens
×Abstract
Major limitations to gene therapy using HSCs are low gene transfer efficiency and the inability of most therapeutic genes to confer a selective advantage on the gene-corrected cells. One approach to enrich for gene-modified cells in vivo is to include in the retroviral vector a drug resistance gene, such as the P140K mutant of the DNA repair enzyme O6-methylguanine-DNA methyltransferase (MGMT*). We transplanted 5 rhesus macaques with CD34+ cells transduced with lentiviral vectors encoding MGMT* and a fluorescent marker, with or without homeobox B4 (HOXB4), a potent stem cell self-renewal gene. Transgene expression and common integration sites in lymphoid and myeloid lineages several months after transplantation confirmed transduction of long-term repopulating HSCs. However, all animals showed only a transient increase in gene-marked lymphoid and myeloid cells after O6-benzylguanine (BG) and temozolomide (TMZ) administration. In 1 animal, cells transduced with MGMT* lentiviral vectors were protected and expanded after multiple courses of BG/TMZ, providing a substantial increase in the maximum tolerated dose of TMZ. Additional cycles of chemotherapy using 1,3-bis-(2-chloroethyl)-1-nitrosourea (BCNU) resulted in similar increases in gene marking levels, but caused high levels of nonhematopoietic toxicity. Inclusion of HOXB4 in the MGMT* vectors resulted in no substantial increase in gene marking or HSC amplification after chemotherapy treatment. Our data therefore suggest that lentivirally mediated gene transfer in transplanted HSCs can provide in vivo chemoprotection of progenitor cells, although selection of long-term repopulating HSCs was not seen.
Authors
Andre Larochelle, Uimook Choi, Yan Shou, Nora Naumann, Natalia A. Loktionova, Joshua R. Clevenger, Allen Krouse, Mark Metzger, Robert E. Donahue, Elizabeth Kang, Clinton Stewart, Derek Persons, Harry L. Malech, Cynthia E. Dunbar, Brian P. Sorrentino
×Abstract
l-Asparaginase is a key therapeutic agent for treatment of childhood acute lymphoblastic leukemia (ALL). There is wide individual variation in pharmacokinetics, and little is known about its metabolism. The mechanisms of therapeutic failure with l-asparaginase remain speculative. Here, we now report that 2 lysosomal cysteine proteases present in lymphoblasts are able to degrade l-asparaginase. Cathepsin B (CTSB), which is produced constitutively by normal and leukemic cells, degraded asparaginase produced by Escherichia coli (ASNase) and Erwinia chrysanthemi. Asparaginyl endopeptidase (AEP), which is overexpressed predominantly in high-risk subsets of ALL, specifically degraded ASNase. AEP thereby destroys ASNase activity and may also potentiate antigen processing, leading to allergic reactions. Using AEP-mediated cleavage sequences, we modeled the effects of the protease on ASNase and created a number of recombinant ASNase products. The N24 residue on the flexible active loop was identified as the primary AEP cleavage site. Sole modification at this site rendered ASNase resistant to AEP cleavage and suggested a key role for the flexible active loop in determining ASNase activity. We therefore propose what we believe to be a novel mechanism of drug resistance to ASNase. Our results may help to identify alternative therapeutic strategies with the potential of further improving outcome in childhood ALL.
Authors
Naina Patel, Shekhar Krishnan, Marc N. Offman, Marcin Krol, Catherine X. Moss, Carly Leighton, Frederik W. van Delft, Mark Holland, JiZhong Liu, Seema Alexander, Clare Dempsey, Hany Ariffin, Monika Essink, Tim O.B. Eden, Colin Watts, Paul A. Bates, Vaskar Saha
×Abstract
Platinum-based drugs that induce DNA damage are commonly used first-line chemotherapy agents for testicular, bladder, head and neck, lung, esophageal, stomach, and ovarian cancers. The inherent resistance of tumors to DNA damage often limits the therapeutic efficacy of these agents, such as cisplatin. An enhanced DNA repair and telomere maintenance response by the Mre11/Rad50/Nbs1 (MRN) complex is critical in driving this chemoresistance. We hypothesized therefore that the targeted impairment of native cellular MRN function could sensitize tumor cells to cisplatin. To test this, we designed what we believe to be a novel dominant-negative adenoviral vector containing a mutant RAD50 gene that significantly downregulated MRN expression and markedly disrupted MRN function in human squamous cell carcinoma cells. A combination of cisplatin and mutant RAD50 therapy produced significant tumor cytotoxicity in vitro, with a corresponding increase in DNA damage and telomere shortening. In cisplatin-resistant human squamous cell cancer xenografts in nude mice, this combination therapy caused dramatic tumor regression with increased apoptosis. Our findings suggest the use of targeted RAD50 disruption as what we believe to be a novel chemosensitizing approach for cancer therapy in the context of chemoresistance. This strategy is potentially applicable to several types of malignant tumors that demonstrate chemoresistance and may positively impact the treatment of these patients.
Authors
Waleed M. Abuzeid, Xiaoling Jiang, Guoli Shi, Hui Wang, David Paulson, Koji Araki, David Jungreis, James Carney, Bert W. O’Malley Jr., Daqing Li
×Abstract
Notch signaling is vital for proper cardiovascular development and function in both humans and animal models. Indeed, mutations in either JAGGED or NOTCH cause congenital heart disease in humans and NOTCH mutations are associated with adult valvular disease. Notch typically functions to mediate developmental interactions between adjacent tissues. Here we show that either absence of the Notch ligand Jagged1 or inhibition of Notch signaling in second heart field tissues results in murine aortic arch artery and cardiac anomalies. In mid-gestation, these mutants displayed decreased Fgf8 and Bmp4 expression. Notch inhibition within the second heart field affected the development of neighboring tissues. For example, faulty migration of cardiac neural crest cells and defective endothelial-mesenchymal transition within the outflow tract endocardial cushions were observed. Furthermore, exogenous Fgf8 was sufficient to rescue the defect in endothelial-mesenchymal transition in explant assays of endocardial cushions following Notch inhibition within second heart field derivatives. These data support a model that relates second heart field, neural crest, and endocardial cushion development and suggests that perturbed Notch-Jagged signaling within second heart field progenitors accounts for some forms of congenital and adult cardiac disease.
Authors
Frances A. High, Rajan Jain, Jason Z. Stoller, Nicole B. Antonucci, Min Min Lu, Kathleen M. Loomes, Klaus H. Kaestner, Warren S. Pear, Jonathan A. Epstein
×Abstract
During human embryogenesis, neural crest cells migrate to the anterior chamber of the eye and then differentiate into the inner layers of the cornea, the iridocorneal angle, and the anterior portion of the iris. When proper development does not occur, this causes iridocorneal angle dysgenesis and intraocular pressure (IOP) elevation, which ultimately results in developmental glaucoma. Here, we show that heparan sulfate (HS) deficiency in mouse neural crest cells causes anterior chamber dysgenesis, including corneal endothelium defects, corneal stroma hypoplasia, and iridocorneal angle dysgenesis. These dysfunctions are phenotypes of the human developmental glaucoma, Peters anomaly. In the neural crest cells of mice embryos, disruption of the gene encoding exostosin 1 (Ext1), which is an indispensable enzyme for HS synthesis, resulted in disturbed TGF-β2 signaling. This led to reduced phosphorylation of Smad2 and downregulated expression of forkhead box C1 (Foxc1) and paired-like homeodomain transcription factor 2 (Pitx2), transcription factors that have been identified as the causative genes for developmental glaucoma. Furthermore, impaired interactions between HS and TGF-β2 induced developmental glaucoma, which was manifested as an IOP elevation caused by iridocorneal angle dysgenesis. These findings suggest that HS is necessary for neural crest cells to form the anterior chamber via TGF-β2 signaling. Disturbances of HS synthesis might therefore contribute to the pathology of developmental glaucoma.
Authors
Keiichiro Iwao, Masaru Inatani, Yoshihiro Matsumoto, Minako Ogata-Iwao, Yuji Takihara, Fumitoshi Irie, Yu Yamaguchi, Satoshi Okinami, Hidenobu Tanihara
×Abstract
Pulmonary hypertension (PH) is an unremitting disease defined by a progressive increase in pulmonary vascular resistance leading to right-sided heart failure. Using mice with genetic deletions of caveolin 1 (Cav1) and eNOS (Nos3), we demonstrate here that chronic eNOS activation secondary to loss of caveolin-1 can lead to PH. Consistent with a role for eNOS in the pathogenesis of PH, the pulmonary vascular remodeling and PH phenotype of Cav1–/– mice were absent in Cav1–/–Nos3–/– mice. Further, treatment of Cav1–/– mice with either MnTMPyP (a superoxide scavenger) or l-NAME (a NOS inhibitor) reversed their pulmonary vascular pathology and PH phenotype. Activation of eNOS in Cav1–/– lungs led to the impairment of PKG activity through tyrosine nitration. Moreover, the PH phenotype in Cav1–/– lungs could be rescued by overexpression of PKG-1. The clinical relevance of the data was indicated by the observation that lung tissue from patients with idiopathic pulmonary arterial hypertension demonstrated increased eNOS activation and PKG nitration and reduced caveolin-1 expression. Together, these data show that loss of caveolin-1 leads to hyperactive eNOS and subsequent tyrosine nitration–dependent impairment of PKG activity, which results in PH. Thus, targeting of PKG nitration represents a potential novel therapeutic strategy for the treatment of PH.
Authors
You-Yang Zhao, Yidan D. Zhao, Muhammad K. Mirza, Julia H. Huang, Hari-Hara S.K. Potula, Steven M. Vogel, Viktor Brovkovych, Jason X.-J. Yuan, John Wharton, Asrar B. Malik
×Abstract
Cardiac atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) modulate blood pressure and volume by activation of the receptor guanylyl cyclase–A (GC-A) and subsequent intracellular cGMP formation. Here we report what we believe to be a novel function of these peptides as paracrine regulators of vascular regeneration. In mice with systemic deletion of the GC-A gene, vascular regeneration in response to critical hind limb ischemia was severely impaired. Similar attenuation of ischemic angiogenesis was observed in mice with conditional, endothelial cell–restricted GC-A deletion (here termed EC GC-A KO mice). In contrast, smooth muscle cell–restricted GC-A ablation did not affect ischemic neovascularization. Immunohistochemistry and RT-PCR revealed BNP expression in activated satellite cells within the ischemic muscle, suggesting that local BNP elicits protective endothelial effects. Since within the heart, BNP is mainly induced in cardiomyocytes by mechanical load, we investigated whether the natriuretic peptide/GC-A system also regulates angiogenesis accompanying load-induced cardiac hypertrophy. EC GC-A KO hearts showed diminished angiogenesis, mild fibrosis, and diastolic dysfunction. In vitro BNP/GC-A stimulated proliferation and migration of cultured microvascular endothelia by activating cGMP-dependent protein kinase I and phosphorylating vasodilator-stimulated phosphoprotein and p38 MAPK. We therefore conclude that BNP, produced by activated satellite cells within ischemic skeletal muscle or by cardiomyocytes in response to pressure load, regulates the regeneration of neighboring endothelia via GC-A. This paracrine communication might be critically involved in coordinating muscle regeneration/hypertrophy and angiogenesis.
Authors
Michaela Kuhn, Katharina Völker, Kristine Schwarz, Javier Carbajo-Lozoya, Ulrich Flögel, Christoph Jacoby, Jörg Stypmann, Martin van Eickels, Stepan Gambaryan, Michael Hartmann, Matthias Werner, Thomas Wieland, Jürgen Schrader, Hideo A. Baba
×Abstract
Chronic prostatitis is a common disease of unclear etiology and has no specific treatment. Mice deficient in the expression of the autoimmune regulator (Aire) gene, which are defective in thymic expression of self antigens and central tolerance, develop spontaneous prostatitis. In this study, we found that Aire-deficient mice developed spontaneous B and T cell immune responses to a prostate autoantigen, seminal vesicle secretory protein 2 (SVS2), which we believe to be novel. We show that thymic expression of this self antigen was Aire dependent. Moreover, prostatitis was induced in WT mice through immunization with SVS2, demonstrating that immunity to SVS2 was sufficient to induce prostatitis. The clinical relevance of this antigen was highlighted by our observation that patients with chronic prostatitis possessed specific autoantibodies against the human SVS2-like seminal vesicle protein semenogelin. These results provide direct evidence that spontaneous chronic prostatitis is an autoimmune disease and is regulated by both central and peripheral tolerance. Moreover, SVS2 and semenogelin are among the relevant autoantigens in mice and humans, respectively.
Authors
Yafei Hou, Jason DeVoss, Vinh Dao, Serena Kwek, Jeffrey P. Simko, Douglas G. McNeel, Mark S. Anderson, Lawrence Fong
×Abstract
Paraneoplastic neurologic disorders (PNDs) offer an uncommon opportunity to study human tumor immunity and autoimmunity. In small cell lung cancer (SCLC), expression of the HuD neuronal antigen is thought to lead to immune recognition, suppression of tumor growth, and, in a subset of patients, triggering of the Hu paraneoplastic neurologic syndrome. Antigen-specific CTLs believed to contribute to disease pathophysiology were described 10 years ago in paraneoplastic cerebellar degeneration. Despite parallel efforts, similar cells have not been defined in Hu patients. Here, we have identified HuD-specific T cells in Hu patients and provided an explanation for why their detection has been elusive. Different Hu patients harbored 1 of 2 kinds of HuD-specific CD8+ T cells: classical IFN-γ–producing CTLs or unusual T cells that produced type 2 cytokines, most prominently IL-13 and IL-5, and lacked cytolytic activity. Further, we found evidence that SCLC tumor cells produced type 2 cytokines and that these cytokines trigger naive CD8+ T cells to adopt the atypical type 2 phenotype. These observations demonstrate the presence of an unusual noncytotoxic CD8+ T cell in patients with the Hu paraneoplastic syndrome and suggest that SCLC may evade tumor immune surveillance by skewing tumor antigen–specific T cells to this unusual noncytolytic phenotype.
Authors
Wendy K. Roberts, Ilana J. Deluca, Ashby Thomas, John Fak, Travis Williams, Noreen Buckley, Athanasios G. Dousmanis, Jerome B. Posner, Robert B. Darnell
×Abstract
Phase II clinical trials revealed that the lymphocyte-depleting humanized monoclonal antibody alemtuzumab (Campath-1H) is highly effective in the treatment of early relapsing-remitting multiple sclerosis. However, 30% of patients develop autoimmunity months to years after pulsed exposure to alemtuzumab, usually targeting the thyroid gland and, more rarely, blood components. In this study, we show that autoimmunity arose in those patients with greater T cell apoptosis and cell cycling in response to alemtuzumab-induced lymphocyte depletion, a phenomenon that is driven by higher levels of IL-21. Before treatment, patients who went on to develop secondary autoimmunity had more than 2-fold greater levels of serum IL-21 than the nonautoimmune group. We suggest that serum IL-21 may, therefore, serve as a biomarker for the risk of developing autoimmunity months to years after alemtuzumab treatment. This has implications for counseling those patients with multiple sclerosis who are considering lymphocyte-depleting therapy with alemtuzumab. Finally, we demonstrate through genotyping that IL-21 expression is genetically predetermined. We propose that, by driving cycles of T cell expansion and apoptosis to excess, IL-21 increases the stochastic opportunities for T cells to encounter self antigen and, hence, for autoimmunity.
Authors
Joanne L. Jones, Chia-Ling Phuah, Amanda L. Cox, Sara A. Thompson, Maria Ban, Jacqueline Shawcross, Amie Walton, Stephen J. Sawcer, Alastair Compston, Alasdair J. Coles
×Abstract
Embryo implantation induces formation of the decidua, a stromal cell–derived structure that encases the fetus and placenta. Using the mouse as a model organism, we have found that this tissue reaction prevents DCs stationed at the maternal/fetal interface from migrating to the lymphatic vessels of the uterus and thus reaching the draining lymph nodes. Strikingly, decidual DCs remained immobile even after being stimulated with LPS and exhibiting responsiveness to CCL21, the chemokine that drives DC entry into lymphatic vessels. An analysis of maternal T cell reactivity toward a surrogate fetal/placental antigen furthermore revealed that regional T cell responses toward the fetus and placenta were driven by passive antigen transport and thus the tolerogenic mode of antigen presentation that predominates when there is negligible input from tissue-resident DCs. Indeed, the lack of involvement of tissue-resident DCs in the T cell response to the fetal allograft starkly contrasts with their prominent role in organ transplant rejection. Our results suggest that DC entrapment within the decidua minimizes immunogenic T cell exposure to fetal/placental antigens and raise the possibility that impaired development or function of the human decidua, which unlike that of the mouse contains lymphatic vessels, might lead to pathological T cell activation during pregnancy.
Authors
Mary K. Collins, Chin-Siean Tay, Adrian Erlebacher
×Abstract
The mammalian epididymis provides sperm with an environment that promotes their maturation and protects them from external stresses. For example, it harbors an array of antioxidants, including non-conventional glutathione peroxidase 5 (GPX5), to protect them from oxidative stress. To explore the role of GPX5 in the epididymis, we generated mice that lack epididymal expression of the enzyme. Histological analyses of Gpx5–/– epididymides and sperm cells revealed no obvious defects. Furthermore, there were no apparent differences in the fertilization rate of sexually mature Gpx5–/– male mice compared with WT male mice. However, a higher incidence of miscarriages and developmental defects were observed when WT female mice were mated with Gpx5-deficient males over 1 year old compared with WT males of the same age. Flow cytometric analysis of spermatozoa recovered from Gpx5-null and WT male mice revealed that sperm DNA compaction was substantially lower in the cauda epididymides of Gpx5-null animals and that they suffered from DNA oxidative attacks. Real-time PCR analysis of enzymatic scavengers expressed in the mouse epididymis indicated that the cauda epididymidis epithelium of Gpx5-null male mice mounted an antioxidant response to cope with an excess of ROS. These observations suggest that GPX5 is a potent antioxidant scavenger in the luminal compartment of the mouse cauda epididymidis that protects spermatozoa from oxidative injuries that could compromise their integrity and, consequently, embryo viability.
Authors
Eléonore Chabory, Christelle Damon, Alain Lenoir, Gary Kauselmann, Hedrun Kern, Branko Zevnik, Catherine Garrel, Fabrice Saez, Rémi Cadet, Joelle Henry-Berger, Michael Schoor, Ulrich Gottwald, Ursula Habenicht, Joël R. Drevet, Patrick Vernet
×Abstract
Liver sinusoidal endothelial cells are a major endogenous source of Factor VIII (FVIII), lack of which causes the human congenital bleeding disorder hemophilia A. Despite extensive efforts, gene therapy using viral vectors has shown little success in clinical hemophilia trials. Here we achieved cell type–specific gene targeting using hyaluronan- and asialoorosomucoid-coated nanocapsules, generated using dispersion atomization, to direct genes to liver sinusoidal endothelial cells and hepatocytes, respectively. To highlight the therapeutic potential of this approach, we encapsulated Sleeping Beauty transposon expressing the B domain–deleted canine FVIII in cis with Sleeping Beauty transposase in hyaluronan nanocapsules and injected them intravenously into hemophilia A mice. The treated mice exhibited activated partial thromboplastin times that were comparable to those of wild-type mice at 5 and 50 weeks and substantially shorter than those of untreated controls at the same time points. Further, plasma FVIII activity in the treated hemophilia A mice was nearly identical to that in wild-type mice through 50 weeks, while untreated hemophilia A mice exhibited no detectable FVIII activity. Thus, Sleeping Beauty transposon targeted to liver sinusoidal endothelial cells provided long-term expression of FVIII, without apparent antibody formation, and improved the phenotype of hemophilia A mice.
Authors
Betsy T. Kren, Gretchen M. Unger, Lucas Sjeklocha, Alycia A. Trossen, Vicci Korman, Brenda M. Diethelm-Okita, Mark T. Reding, Clifford J. Steer
×Abstract
Dorsal root ganglion (DRG) neuron dysfunction occurs in a variety of sensory neuronopathies for which there are currently no satisfactory treatments. Here we describe the development of a strategy to target therapeutic genes to DRG neurons for the treatment of these disorders. We genetically modified an adenovirus (Ad) to generate a helper virus (HV) that was detargeted for native adenoviral tropism and contained DRG homing peptides in the adenoviral capsid fiber protein; we used this HV to generate DRG-targeted helper-dependent Ad (HDAd). In mice, intrathecal injection of this HDAd produced a 100-fold higher transduction of DRG neurons and a markedly attenuated inflammatory response compared with unmodified HDAd. We also injected HDAd encoding the β subunit of β-hexosaminidase (Hexb) into Hexb-deficient mice, a model of the neuronopathy Sandhoff disease. Delivery of the DRG-targeted HDAd reinstated neuron-specific Hexb production, reversed gangliosidosis, and ameliorated peripheral sensory dysfunction. The development of DRG neuron–targeted HDAd with proven efficacy in a preclinical model may have implications for the treatment of sensory neuronopathies of diverse etiologies.
Authors
Tomoya Terashima, Kazuhiro Oka, Angelika B. Kritz, Hideto Kojima, Andrew H. Baker, Lawrence Chan


























Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)