Research Article

Abstract
Single-agent anti-PD-1 antibodies are ineffective for pancreatic ductal adenocarcinoma (PDAC) due to the immunosuppressive tumor-microenvironment (TME). KRAS mutations contribute to the inflammatory TME and therapeutic resistance by upregulating IL-8 via MAPK pathways. Thus, this study attempted to overcome the resistance to anti-PD-1 antibodies by targeting downstream KRAS-effectors. The study found that the resistance to anti-PD-1 antibodies can be overcome through MEK1/2-inhibition. The combination of anti-PD-1 antibodies and MEK inhibitors displayed antitumor activity in Kras mutated (Krasmut) KPC mouse tumors, but not WT (KrasWT) Panc02 tumors. The combination of anti-PD-1 antibodies and MEK inhibitors induced recruitment of tumor-associated neutrophils (TANs) via CXCR2, an IL-8 receptor, and increased memory CD8+ T cells and IFN-γ production in treatment-sensitive tumors. However, larger tumors still resisted the combination of anti-PD-1 antibody and MEK inhibitor, likely due to hypoxia/necrosis-induced NETosis and associated paucity of CD8+ T cells. The subsequent addition of anti-CXCR2 antibody overcame this resistance by blocking TAN-infiltration to hypoxic/necrotic areas. Consistently, a risk-score based on the NETosis-MAPK signaling interaction is significantly associated with poorer survival in human PDAC. This study thus provides a new venue for overcoming resistance to strategies targeting KRAS signaling.
Authors
Brian Herbst, Alex Blair, Yiming Li, Elizabeth M. Jaffee, Lei Zheng

Abstract
Epigenetic dysregulation is associated with immune evasion and immune checkpoint blockade (ICB) resistance. Here, using in vivo CRISPR/Cas9 screens targeting epigenetics-related factors in mouse tumor models treated with ICB, we identified chromobox 4 (CBX4) as a key negative regulator of the immune tumor microenvironment (TME). Single-cell RNA-seq and spatial transcriptomics analyses of patients receiving neoadjuvant anti–programmed cell death protein 1 (anti–PD-1) therapy revealed high CBX4 expression in both tumor cells and immunosuppressive tumor-associated macrophage subpopulations, with preferential accumulation in nonresponders. Deficiency of CBX4 in macrophages or tumor cells induced robust antitumor immunity and increased infiltration and the cytotoxic activity of CD8+ T cells and NK cells, thereby heightening the sensitivity of ICB treatment. Mechanistically, CBX4 targeted H3K9me3- and H3K27me3-marked endogenous retroelements such as RLTR4-Mm-int. Loss of CBX4 derepressed retrotransposons, activating cytosolic RNA-sensing pathways and triggering the type I IFN response, ultimately leading to a robustly inflamed TME. Moreover, we uncovered a negative correlation between CBX4 expression, immune responses, and retrotransposon levels, and were able to determine the prognosis of patients with hepatocellular carcinoma (HCC) undergoing ICB therapy. Our study establishes CBX4 as an epigenetic immune checkpoint through the epigenetic silencing of retrotransposons, remodeling the immune TME and thus providing a promising therapeutic target to enhance tumor immunogenicity and overcome immunotherapy resistance.
Authors
Zhibo Ma, Wenlong Jia, Xi Zhou, Jing Liu, Qingwen Li, Ruizhi Chang, Gu Shiqi, Naonao Yuan, Zhishui Chen, Peixiang Lan

Abstract
Understanding susceptibility factors of sepsis is crucial for early diagnosis and development of personalized treatment strategies. However, the genetic determinants for initiation and progression of sepsis remain unclear. Here, we showed that the expression levels of estrogen receptor β (ERβ) were significantly reduced in the peripheral blood of patients with sepsis and were negatively correlated with disease severity. The results from human samples and experimental animals demonstrated that ERβ deficiency enhanced the body’s susceptibility to sepsis by inducing macrophage pyroptosis, thereby impairing bacterial clearance. Mechanistically, ERβ deficiency enhanced fatty acid oxidation, increased acetyl-CoA levels, and promoted acetylation of stomatin-like protein 2 (Stoml2) at K221, leading to mitochondrial dysfunction and macrophage pyroptosis. Mutating the Stoml2 K221 site mitigated these effects and improved survival of septic mice. These findings suggest ERβ deficiency as a potential genetic factor in sepsis susceptibility.
Authors
Yanrong Zhu, Gang Li, Yilei Guo, Yue He, Wanyi Zhang, Lei Gao, Jing Zhang, Pengxiang Guo, Haochang Lin, Wenjie Zhang, Zhifeng Wei, Yufeng Xia, Yue Dai
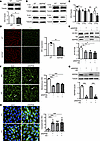
Abstract
Diabetic retinopathy involves early retinal vascular barrier breakdown and pericyte loss, yet the initiating molecular events remain poorly defined. Vascular endothelial cadherin (VE-cadherin), a key regulator of endothelial integrity, is notably reduced in diabetic and prediabetic nucleoside diphosphate kinase B–deficient (NDPKB-deficient) mouse retinas, particularly in the retinal deep capillary layer, and this decline precedes pericyte loss. In vitro, high glucose (HG) and NDPKB deficiency induced VE-cadherin Y685 phosphorylation, promoting its junctional internalization, activating the hexosamine biosynthesis pathway, and increasing angiopoietin 2 (Ang2), resulting in impaired endothelial barrier function and disrupting pericyte attachment. Preventing Y685 phosphorylation through VE-cadherin Y685F mutation blocked these HG- and NDPKB-driven pathological effects. Pharmacological intervention experiments identified protein O-linked β-N-acetyl glucosamine (O-GlcNAc) modification as a mediator of Y685-dependent Ang2 upregulation. In vivo, VE-cadherin Y685F-knockin mice were protected from diabetes- and prediabetes-induced vascular hyperpermeability, exhibited reduced protein O-GlcNAcylation and Ang2 induction, and maintained neuronal function. O-GlcNAc–enriched retinal proteomics further showed that the Y685F mutation restored balanced neurovascular and mitochondrial pathways. These findings highlight the potential of targeting VE-cadherin Y685 phosphorylation as a promising therapeutic approach to maintain retinal vascular integrity and attenuate the pathological progression of diabetic and prediabetic retinopathy.
Authors
Yixin Wang, Hongpeng Huang, Feng Shao, Rachana Eshwaran, Miao Qin, Noor Karim, Yonggang Ren, Gergana Dobreva, Hans-Peter Hammes, Thomas Wieland, Yuxi Feng

Abstract
Cachexia is a metabolic wasting syndrome affecting many patients with cancer, with poor survival outcomes. Disturbed lipid metabolism is a hallmark of cachexia, and our previous work has identified increased levels of circulating ceramides, which are bioactive lipids with adverse effects in metabolic diseases, as biomarkers for cachexia in mouse models and patients. Here, we investigated the role of ceramides on cachexia development using the well-established C26 colon carcinoma model. We demonstrated that elevated ceramides in cachexia arose from increased liver synthesis. We showed that ceramides directly contributed to impaired mitochondrial function and energy homeostasis in cachexia target tissues. Targeting ceramide synthesis using miRNA interference, or myriocin, an approved compound targeting the key synthesis enzyme serine palmitoyltransferase (SPT), improved markers of muscle atrophy in cachectic male mice. Importantly, we demonstrated that key enzymes involved in ceramide production were also elevated in livers, but not in other organs, of patients with cancer cachexia, correlating with disease severity. Our data place ceramides as contributors to metabolic dysfunction in cachexia and highlight the suitability of the ceramide synthesis pathway for therapeutic targeting.
Authors
Pauline Morigny, Honglei Ji, Laura Cussonneau, Sabrina Zorzato, Yun Kwon, Fabien Riols, Doris Kaltenecker, Alisa Maier, Vignesh Karthikaisamy, Samantha Corrà, Tanja Krauss, Claudine Seeliger, Syed Qaaifah Gillani, Joël J. Tissink, Sandra Lacas-Gervais, Tuna Felix Samanci, Adriano Maida, Raul Terron-Exposito, Angela Trinca, Christine von Toerne, Leonardo Nogara, Melina Claussnitzer, Olga Prokopchuk, Jeannine Bachmann, Mauricio Berriel Diaz, Laure B. Bindels, Ondrej Kuda, Hans Hauner, Mark Haid, Stephan Herzig, Carlo Fiore Viscomi, Jerome Gilleron, Anja Zeigerer, Bert Blaauw, Maria Rohm

Abstract
While clinical trials of human pluripotent stem cell–derived midbrain dopamine (mDA) neuron precursor grafts for Parkinson’s disease (PD) are ongoing, current protocols remain suboptimal. In particular, the yield of TH+ mDA neurons after in vivo grafting and the expression of certain mDA neuron and subtype-specific markers require improvement. Single-cell transcriptomic analyses of grafts have revealed low proportions of mDA neurons and substantial off-target contamination. Here, we present an optimized mDA neuron differentiation strategy that builds on our clinical-grade (“Boost”) protocol by adding FGF18 and IWP2 treatment (“Boost+”) at the neurogenesis stage. Boost+ mDA neurons show higher expression of EN1, PITX3, and ALDH1A1. Improvements in mDA neuron yield and transcriptional similarity to primary mDA neurons are observed in vitro and following transplantation. Single-nucleus RNA sequencing demonstrates enrichment of A9 mDA neurons within Boost+ grafts. Functional studies in vitro demonstrate increased dopamine production and release and improved electrophysiological properties. In vivo analyses show higher percentages of TH+ mDA neurons, resulting in efficient rescue of amphetamine-induced rotation behavior in the 6-OHDA rat model and rescue of deficits in some nondrug-induced assays, including the ladder rung assay, which are not improved by Boost mDA neurons. The Boost+ conditions present an optimized differentiation protocol with advantages for disease modeling and mDA neuron grafting paradigms.
Authors
Tae Wan Kim, Jinghua Piao, Vittoria D. Bocchi, So Yeon Koo, Se Joon Choi, Fayzan Chaudhry, Donghe Yang, Hyein S. Cho, Emiliano Hergenreder, Lucia Ruiz Perera, Subhashini Joshi, Zaki Abou Mrad, Nidia Claros, Shkurte Ademi Donohue, Yeong Eun Im, Hyo Jae Jeong, Anika K. Frank, Ryan M. Walsh, Eugene V. Mosharov, Doron Betel, Viviane Tabar, Lorenz Studer

Abstract
Skraban-Deardorff syndrome, a rare neurodevelopmental disorder caused by WD repeat domain 26 (WDR26) haploinsufficiency, is characterized by intellectual disability, seizures, autistic-like behaviors, and craniofacial anomalies. Despite its genetic association with variants disrupting the C-terminal to LisH (CTLH) E3 ubiquitin ligase complex, the molecular mechanisms linking WDR26 dysfunction to neurodevelopmental deficits remain unclear. Here, we demonstrate that Wdr26 heterozygous-KO mice (Wdr26+/–) recapitulated core clinical features of the syndrome, including learning and memory impairments, social dysfunction, heightened seizure susceptibility, and motor deficits, alongside rare craniofacial and dental abnormalities. Mechanistically, Wdr26 haploinsufficiency stabilized RUNX1 translocation partner 1 (RUNX1T1), a transcriptional coactivator critical for neuronal differentiation, by impairing its ubiquitination and proteasomal degradation, consequently disrupting the level of microtubule-associated protein 2 (MAP2), a key regulator of dendritic architecture and synaptic plasticity. Early intervention in neonatal Wdr26+/– mice (P0.5) using AAV-shRNA–mediated Runx1t1 knockdown reversed MAP2 overexpression and behavioral deficits. Notably, the antipsychotic risperidone ameliorated cognitive and social impairments in Wdr26+/– mice by upregulating WDR26 levels, suggesting a potential therapeutic avenue. Our findings not only establish the animal model as a robust preclinical tool but also define the WDR26/RUNX1T1/MAP2 regulatory axis as pivotal to the syndrome’s pathogenesis, while identifying actionable therapeutic targets.
Authors
Xingyun Xu, Yaohui Zhou, Shiyao Xu, Hongjie Zhou, Xuexia Lin, Yuhao Luo, Yu Xu, Zhigang Miao, Wei Ge, Hao Yang, Xingshun Xu
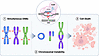
Abstract
While radiation is an effective oncologic therapy, killing cancer by inducing DNA double-strand breaks (DSBs), it lacks specificity for neoplastic cells. We have previously adapted the CRISPR/Cas9 gene-editing technology as a cancer-specific treatment modality targeting somatic mutations in pancreatic cancer (PC). However, its tumoricidal potential remains unclear, especially in comparison with therapeutic doses of radiation. Here, we demonstrate that CRISPR/Cas9-induced DSBs are more cytotoxic in PCs than a comparable number of radiation-induced DSBs. We observed more than 90% tumor growth inhibition by targeting 9 sites with cancer-specific sgRNAs. Through both bioinformatics and cytogenetics analyses, we found that CRISPR/Cas9-induced DSBs triggered ongoing chromosomal rearrangements, with 87% of structural variants not directly produced from the initial CRISPR/Cas9-induced DSBs, and chromosomal instability peaking before cell death. By comparing the cytotoxicity of CRISPR/Cas9- and radiation-induced DSBs, we demonstrated that the number of DSBs required to achieve equitoxic effects was approximately 3 times higher for radiation than CRISPR/Cas9. Finally, we showed that PC cells that had survived CRISPR/Cas9 targeting retained susceptibility to subsequent CRISPR/Cas9-induced DSBs at different genomic sites with more than 87% growth inhibition. Together, our data support the therapeutic potential of CRISPR/Cas9 as an anticancer strategy.
Authors
Selina Shiqing K. Teh, Akhil Kotwal, Alexis Bennett, Eitan Halper-Stromberg, Laura Morsberger, Saum Zamani, Yanan Shi, Alyza Skaist, Qingfeng Zhu, Kirsten Bowland, Hong Liang, Ralph H. Hruban, Chien-Fu Hung, Robert A. Anders, Nicholas J. Roberts, Robert B. Scharpf, Michael Goldstein, Ying S. Zou, James R. Eshleman
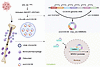
Abstract
Bone metastasis remains a major cause of morbidity in estrogen receptor–positive breast cancer, with RANKL inhibitor resistance emerging as a critical clinical challenge. Nearly 40% of patients develop progressive skeletal lesions despite denosumab therapy, highlighting an urgent need to identify resistance mechanisms and alternative therapeutic strategies. We identified a RANKL-independent osteoclast activation pathway mediated by the CRKL/circCCDC50/NFATc1 axis. Mechanistically, CRKL promoted EIF4A3-dependent circCCDC50 biogenesis, which was packaged into large oncosomes and transferred to osteoclast precursors. Nuclear circCCDC50 recruited CARM1 to epigenetically activate NFATc1 transcription, establishing a self-reinforcing loop that sustained osteolysis despite RANKL blockade. Pharmacological inhibition of CARM1 (TP-064) effectively suppressed osteoclastogenesis and bone metastasis in denosumab-resistant models. These findings revealed a targetable resistance mechanism and provided a clinically actionable strategy to overcome microenvironment-driven metastasis through dual targeting of tumor and bone niches.
Authors
Qun Lin, Jinpeng Luo, Zhuxi Duan, Jieer Luo, Wei Zhang, Yuan Xia, Yinduo Zeng, Xiaolin Fang, Jiahui Liang, Jiayi Chen, Qianchong Lin, Yilin Quan, Ruiyu Hu, Hongcai Liu, Qiang Liu, Jun Li, Chang Gong

Abstract
Stem cells are critical for the homeostasis of adult tissues. Thyroid hormone (TH), whose intracellular concentration is increased by type 2 deiodinase (D2), is involved in many functions, but its role in quiescence is unknown. Here, we show that D2 marks quiescent stem cells in muscle and skin. Genetic D2 depletion in quiescent muscle stem cells triggered their transition from a G0 to a GAlert-like state. This increased the proliferative potential of the stem cells but impaired their self-renewal capacity, leading to depletion of the stem cell pool and regenerative failure over time. Mechanistically, TH sustained Notch signaling, and active Notch overexpression partially rescued D2 depletion. Transient pharmacological inhibition of D2 accelerated muscle regeneration and skin wound healing by promoting stem cell expansion. In conclusion, we show that D2 is a critical metabolic enzyme in maintaining stem cell quiescence and in regulating self-renewal.
Authors
Maria Angela De Stefano, Raffaele Ambrosio, Cristina Luongo, Tommaso Porcelli, Daniela Di Girolamo, Caterina Miro, Monica Dentice, Caterina Missero, Domenico Salvatore
No posts were found with this tag.



Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)