Neuroscience

Abstract
Enteric nervous system (ENS) injury, characterized by progressive degeneration of enteric neurons and glial cells, is a common diabetic complication with no effective cure beyond symptomatic management. Enteric neural precursor cells (ENPCs) play a key role in maintaining neurogenesis and gliogenesis within the adult ENS. Here, we demonstrate that bone marrow mesenchymal stem cell–derived microvesicles (BMSC-MVs) alleviate diabetic ENS injury. In both diabetic patients and mouse models, gastrointestinal transit was delayed, ENS structure was impaired, and neurogenesis and gliogenesis from ENPCs were elevated yet remained functionally insufficient. Transcriptomic profiling revealed activation of ER stress and the pro-apoptotic PERK branch of the unfolded protein response in ENPCs. BMSC-MVs homed to the colon, were internalized by ENPCs, and suppressed ER stress, thereby enhancing functional neurogenesis and gliogenesis, restoring ENS structure, and improving gastrointestinal motility. Mechanistically, vinculin on BMSC-MVs bound talin-1 on ENPCs, activating the ERK pathway to suppress diabetic ER stress. These results identify BMSC-MVs as a promising cell-free therapeutic strategy for diabetic ENS injury.
Authors
Huiying Shi, Hailing Yao, Yilin Liu, Mengke Fan, Sicheng Cai, Shizhao Xu, Chen Jiang, Yurui Zhang, Weiwei Jiang, Wei Qian, Rong Lin
Abstract
Huntington’s disease (HD) is a fatal neurodegenerative disorder characterized by progressive motor dysfunction, cognitive decline, and striatal neuron degeneration, primarily affecting medium spiny neurons (MSNs). Despite extensive research, the underlying metabolic vulnerabilities contributing to HD pathogenesis remain poorly understood. In this study, we employ RNA sequencing (RNA-seq) and metabolomics analyses to identify marked dysregulation of one-carbon metabolism in HD. We validate that SHMT2, a key mitochondrial enzyme in the mitochondrial one-carbon (mt-1C) pathway, is substantially downregulated in HD patient-derived iPSC-differentiated human striatal organoids (hSOs) and YAC128 mice. Functionally, pharmacological inhibition or genetic deletion of SHMT2 exacerbates mutant huntingtin (mHTT) aggregation, induces MSN degeneration in hSOs, and impairs motor function in WT mice. Conversely, SHMT2 overexpression attenuates MSN degeneration in HD-hSOs and improves motor performance in YAC128 mice. Mechanistically, SHMT2 deficiency leads to homocysteine (HCY) accumulation, which interacts with AARS1 and suppresses histone lactylation, thereby perturbing transcriptional regulation and associating with neurodegenerative phenotypes. Finally, we demonstrate that the HD clinical drug haloperidol modulates SHMT2 expression and restores histone lactylation, providing a pharmacological tool to probe SHMT2-dependent metabolic and epigenetic regulation in HD models. These findings highlight a metabolic-epigenetic axis as a promising therapeutic target for HD.
Authors
Mingqin Lu, Kexin Li, Shanshan Wu, Zhilong Zheng, Xinyue Li, Shengda Wang, Hanwen Yu, Chunyue Liu, Yueqing Jiang, Xueqin Song, Yan Liu, Xing Guo
Abstract
The mammalian brain relies primarily on glucose for its energy needs. Delivery of this nutrient to the brain is mediated by the glucose transporter-1 (GLUT1) protein. Low GLUT1 thwarts glucose entry into the brain, causing an energy crisis and, triggering, in one instance, the debilitating neurodevelopmental condition – GLUT1 deficiency syndrome (GLUT1DS). Current treatments for GLUT1DS are sub-optimal, as none address the root cause – low GLUT1 – of the condition. Levels of this transporter must respond rapidly to the brain’s changing energy requirements. This necessitates fine-tuning its expression. Here we describe a long-noncoding RNA (lncRNA) antisense to GLUT1 (SLC2A1) and show that it is involved in such regulation. Raising levels of the lncRNA had a concordant effect on GLUT1 in cultured human cells and transgenic mice; reducing levels elicited the opposite effect. Delivering the lncRNA to GLUT1DS model mice via viral vectors induced GLUT1 expression, enhancing brain glucose levels to mitigate disease. Direct delivery of such a lncRNA to combat disease has not been reported previously and constitutes, to our knowledge, a unique therapeutic paradigm. Moreover, considering the importance of maintaining homeostatic GLUT1 levels, calibrating transporter expression via the lncRNA could become broadly relevant to myriad conditions, including Alzheimer’s disease, wherein GLUT1 is perturbed.
Authors
Maoxue Tang, Sasa Teng, Yueqing Peng, Ashley Y. Kim, Yoon-Ra Her, Peter Canoll, Jeffrey N. Bruce, Phyllis L. Faust, Kailash Adhikari, Darryl C. De Vivo, Umrao R. Monani
Abstract
BACKGROUND. Estrogen deficiency and progressive hearing loss (HL) are significant concerns in individuals with Turner syndrome (TS). However, whether childhood estrogen deficiency increases HL risk and whether estrogen replacement therapy (ERT) prevents hearing deterioration are still unclear. METHODS. This prospective cohort study recruited children with TS from a tertiary referral center between 2016 and 2024. All participants received standardized recombinant human growth hormone therapy. Longitudinal monitoring data of hormone levels, metabolic parameters, and annual audiological examinations were recorded. The primary analysis used a multivariate Cox model to estimate the adjusted hazard ratio of hearing loss between estrogen-deficient and estrogen-normal TS patients without prior exogenous estrogen exposure. The secondary analysis compared annual pure tone average (PTA) and its changes between the ERT and non-ERT groups in a substudy. RESULTS. Among 87 prepubertal pediatric TS patients, 48 (55.2%) were estrogen-deficient, 38 HL events occurred over 35-month median follow-up. The estrogen-deficient group had higher HL incidence (27 cases, 56.3%; 20.6/100 person-years [PY]) versus estrogen-normal (11 cases, 28.2%; 8.6/100 PY), with estrogen deficiency independently increasing HL risk (HR = 2.93; 95%CI:1.21–7.12). Notably, estrogen deficiency also independently predicted abnormal DPOAE with an even higher effect size (HR = 3.98, 95% CI: 1.35–11.76). The substudy found that initiating ERT at age of 12 significantly preserve auditory function, with the ERT group showing markedly lower PTA and slower hearing deterioration (–1.24 dB/y vs. 1.13 dB/y right ear; –1.85 dB/y vs. 1.04 dB/y left ear, P = 0.001). CONCLUSION. Childhood estrogen deficiency is a modifiable risk factor. Initiating ERT around early adolescence may help hearing preservation. TRIAL REGISTRATION. ChiCTR2300068063. FUNDING. National Natural Science Foundation of China (82173154 and 82471155), Fundamental Research Funds for the Central Universities, Clinical Research 5010 Program, Sun Yat-sen University: 2024004.
Authors
Yan Huang, Liyang Liang, Yanfang Ye, Lina Zhang, Li Ling, Zhe Meng, Wei Liu, Jia Guo, Zulin Liu, Zhen Zhao, Zhigang Zhang, Yu Si

Abstract
N-acetyl-l-leucine (NALL), a derivative of the branched-chain amino acid leucine, has shown therapeutic potential for neurodegenerative diseases, including in prodromal stages of Parkinson’s disease (PD). However, the mechanism of its protective effects has been largely unknown. Using human induced pluripotent stem cell–derived dopaminergic neurons from patients carrying GBA1, LRRK2, or VPS35 mutations, as well as from sporadic PD cases, we found that NALL treatment markedly reduced Ser129 phosphorylated α-synuclein (pS129-syn). Discovery-based proteomic analysis revealed that NALL treatment upregulated lysosomal, mitochondrial, and synaptic proteins without inducing cytotoxicity. The reduction of pS129-syn was dependent on serine protease HTRA1, which was robustly induced by NALL. Moreover, NALL increased the expression of wild-type parkin in mutant dopaminergic neurons, leading to increased glycosylated dopamine transporter, elevated synaptic membrane-associated synaptojanin-1, and accelerated synaptic vesicle endocytosis, suggesting improved synaptic function. Furthermore, in LRRK2R1441C knockin mice, NALL administration decreased pS129-syn, elevated parkin levels, and ameliorated dopamine-dependent motor learning deficits. These findings highlight the therapeutic potential of NALL for PD by its protective effects on α-synuclein pathology and synaptic function in vulnerable dopaminergic neurons.
Authors
Pingping Song, Chuyu Chen, Rossella Franchini, Bryan Duong, Yi-Zhi Wang, Robert Coukos, Zhong Xie, Jeffrey N. Savas, Yueqin Zhou, Mariarita Bertoldi, D. James Surmeier, Loukia Parisiadou, Dimitri Krainc
Abstract
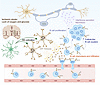
Neuroinflammation, encompassing both innate and adaptive immune responses, plays a crucial role in ischemic stroke. Although B lymphocytes are central to adaptive immunity, their contributions to ischemic stroke remain poorly understood. Here, we demonstrated that B lymphocytes accumulate in ischemic lesions, forming germinal center–like structures at the later stage after stroke, which mainly depended on in situ proliferation. This accumulation correlated with worsened neuroinflammation and ischemic injury, whereas B cell depletion reduced chronic brain damage during stroke. Mechanistically, microglia recruited B cells into ischemic lesions through MIF-CD74/CXCR4 signaling during the early phase of stroke, while IFN-related pathways in B cells further drove neuroinflammation and brain injury. Targeting these pathways markedly alleviated cerebral ischemia and inflammation. Our findings shed light on the role of B lymphocytes in stroke pathology and suggest promising new avenues for therapeutic intervention.
Authors
Sheng Yang, Hang Zhang, Lu-Lu Xu, Luo-Qi Zhou, Yun-Hui Chu, Lian Chen, Xiao-Wei Pang, Lu-Yang Zhang, Li-Fang Zhu, Ming-Hao Dong, Ke Shang, Jun Xiao, Long-Jun Wu, Wei Wang, Dai-Shi Tian, Chuan Qin
Abstract
Dominant-inactivating mutations in the colony stimulating factor-1 receptor (CSF1R) cause CSF-1R related leukoencephalopathy (CRL), an adult-onset neurodegenerative disease that is modeled in the Csf1r+/– mouse. CRL is caused by microglial dysfunction. However, the primary microglial deficit, is unknown. To address this question, we employed single-nucleus RNA sequencing of brains from young Csf1r+/– mice without pathological or behavioral alterations. Reduction of CSF-1R signaling caused metal ion accumulation in brain macrophages, with concomitant activation of cell death and stress response pathways in oligodendrocytes and neuronal subpopulations. Reduction of metallothionein 1 (Mt1) and 3 (Mt3) gene expression was a common feature in glial and neuronal cells of Csf1r+/– mice. Overexpression of Mt1 restored metal ion homeostasis, normalized ROS production in microglia, and prevented the development of behavioral deficits, while Mt3 deletion had disease-enhancing effects. These findings demonstrate CSF-1R regulation of metal ion homeostasis via metallothioneins in the brain.
Authors
Violeta Chitu, Julia Alvarenga, Wenna Chen, David Reynolds, Yang Liu, Daqian Sun, Anders Sandell, Virginjia Danylaite Karrenbauer, Per Uvdal, Iran A.N. da Silva, Christophe Sandt, Oxana Klementieva, Ulf Johansson, Kavitha Subramanian Vignesh, Zbigniew K. Wszolek, Dennis W. Dickson, Jennifer Aguilan, Simone Sidoli, Deyou Zheng, E. Richard Stanley
Abstract
Mild traumatic brain injury (mTBI) from closed-head injuries (CHI) can lead to prevalent neuropsychiatric disorders, including mood disorders and an increased risk for neurodegenerative diseases and dementia. Inflammasomes are molecular complexes crucial for neuroinflammation and secondary damage after trauma, however their role in mild CHI is poorly understood. In this study, we investigate the cellular expression of inflammasome-related genes and their functional significance in CHI models. Single-cell RNA sequencing analysis of cortical tissue after trauma revealed selective expression of Asc (also known as Pycard), which encodes the inflammasome adaptor Apoptosis-associated Speck-like protein containing a Caspase recruitment domain (ASC), predominantly in microglial clusters. Sustained upregulation of inflammasome-related proteins, microglia activation and astrocyte reactivity persisted up to 21 days in a model for mTBI, with this pattern significantly reduced in Asc-/- mice. Importantly, mild cognitive impairment induced after mild CHI was largely abrogated in Asc-/- mice. These findings suggest that ASC, as the primary inflammasome adaptor, plays a critical role in sustaining neuroinflammation and contributes to cognitive deficits after mild CHI. This study provides insights into the molecular neuroinflammatory mechanisms underlying CHI, potentially informing future therapeutic strategies.
Authors
Tao Li, Sergio Castro-Gomez, Pablo Botella Lucena, Ana Vieira-Saecker, Stephanie Schwartz, Yingying Ding, Yushuang Deng, Maling Guo, Valentin Stein, Douglas T. Golenbock, Eicke Latz, Michael T. Heneka
Abstract
Hypomorphic variants in the SEL1L-HRD1 ER-associated degradation (ERAD) complex have been linked to severe neurological syndromes in children, including neurodevelopmental delay, intellectual disability, motor dysfunction, and early death. Despite this association, its physiological importance and underlying mechanisms in neurons remain poorly understood. Here, we show that neuronal SEL1L-HRD1 ERAD is essential for maintaining one-carbon metabolism, motor function, and overall viability. Neuron-specific deletion of Sel1L in mice (Sel1LSynCre) resulted in growth retardation, severe motor impairments, and early mortality by 9 weeks of age—mirroring core clinical features observed in affected patients—despite preserved neuronal numbers and only modest ER stress. Multi-omics analyses, including single-nucleus RNA sequencing and metabolomics, revealed significant dysregulation of one-carbon metabolism in ERAD-deficient brains. This included activation of the serine, folate, and methionine pathways, accompanied by elevated levels of S-adenosylmethionine and related metabolites, likely resulted from induction of the integrated stress response (ISR). Together, these findings uncover a previously unappreciated role for neuronal SEL1L-HRD1 ERAD in coordinating ER protein quality control with metabolic adaptation, providing new insight into the molecular basis of ERAD-related neurodevelopmental disease.
Authors
Mauricio Torres, You Lu, Brent Pederson, Hui Wang, Anna Gretzinger, Liangguang Lin, Jiwon Hwang, Xinxin Chen, Alan C. Rupp, Abigail J. Tomlinson, Andrew J. Scott, Zhen Zhao, Daniel R. Wahl, Martin Myers, Jr, Costas A. Lyssiotis, Ling Qi
Abstract
SLC13A5 citrate transporter disorder is a rare epileptic encephalopathy caused by loss of function pathogenic variants in the SLC13A5 gene. Loss of sodium/citrate cotransporter (NaCT) function causes a severe early life epilepsy resulting in life-long developmental disabilities and increased extracellular citrate. Current antiseizure medications may reduce seizure frequency, yet more targeted treatments are needed to address the epileptic and neurodevelopmental SLC13A5 phenotype. We performed preclinical studies in SLC13A5 deficient mice evaluating phenotype rescue with adeno-associated virus (AAV) vector carrying a functional copy of the human SLC13A5 gene (AAV9/SLC13A5). Cerebrospinal fluid-delivery of AAV9/SLC13A5 decreased extracellular citrate levels, normalized electrophysiologic and sleep architecture abnormalities, and restored resistance to chemically induced seizures and death. Treatment benefits were achieved with administration during early brain development and in young adult mice, indicating therapeutic efficacy across developmental and post-developmental stages. Comparison of delivery routes in young adult KO mice showed that higher brain targeting achieved with intra-cisterna magna delivery resulted in greater treatment benefit as compared to intrathecal lumbar puncture delivery. Together, these results support further development of AAV9/SLC13A5 for treating SLC13A5 citrate transporter disorder.
Authors
Lauren E. Bailey, Raegan M. Adams, Morgan K. Schackmuth, Irvin T. Garza, Krishanna Knight, Sydni K. Holmes, Meghan M. Eller, MinJae Lee, Rachel M. Bailey










Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)