Oncology
Abstract
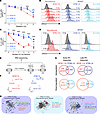
Patients with malignant peripheral nerve sheath tumors (MPNSTs) have poor outcomes despite multimodal treatment with surgery, radiation, and systemic therapy. The responses to radiotherapy (RT) are mixed, and the biologic mechanisms underlying this heterogeneity in the radiation response of MPNSTs are not understood. Here, we combined bulk and single-cell transcriptomics, genome-wide CRISPR interference screens, and multiplatform molecular analysis across MPNST cells, mouse allograft models, and patients’ samples to understand the mediators of the radiation response. Our data revealed that MPNSTs, but not benign plexiform neurofibromas, induced a type I IFN signature that functionally mediated the radiation response. Moreover, irradiation of immunocompetent mouse MPNST allografts led to IFN-mediated T cell recruitment and activation. Both host mouse T cells and intact tumor IFN receptor signaling were required for RT’s efficacy in mouse MPNST allografts. Analysis of human MPNST resection specimens demonstrated that increased microenvironmental and CD8+ T cell infiltration were associated with improved local control following RT. These results provide a preclinical rationale for combining immunomodulatory agents targeting IFN signaling to improve radiation responses in MPNSTs and potentially other soft tissue sarcomas.
Authors
Iowis Zhu, Julian Chien, Gabriel E. Rech, Kanish Mirchia, Sixuan Pan, Kaeli Miller, Joanna Pak, Rosanna Wustrack, Varun Monga, Steve E. Braunstein, Mark D. Adams, Line Jacques, Melike Pekmezci, S. John Liu, Harish N. Vasudevan

Abstract
Liver invasion is one of the most frequent events in the progression of gallbladder cancer (GBC). However, the cellular and pathological role of the tumor-liver–interface microenvironment in liver invasion is still enigmatic. Here, we applied single-cell and spatial transcriptomics to systematically investigate the cellular component and gene expression regulation of the microenvironment from the tumor to the liver, specifically the invasive boundary. Our analyses revealed that CXCL9+ macrophage–rich immune cell niches were accumulated in the tumor-liver invasive margin, where 2 subclasses of the CXCL9+ immune cell niches, CXCL9+TRAC+ (CT) and CXCL9+C1QB+ (CC) niches, were identified. CD8+ T cells were recruited by CXCL9+ macrophages through CXCL9-CXCR3 interaction in the CT niche, which was located adjacent to the liver. Moreover, the CC niche was proximal to the tumor core, where tumor cells induced CD8+ T cell exhaustion via LGALS4 expression. In addition, our cohort study showed that high CXCL9 and low LGALS4 in the liver invasion margin demonstrated a favorable prognosis and better responses to anti–PD-1 immunotherapy for patients with gallbladder cancer. Altogether, these findings demonstrate novel cellular and molecular mechanisms underlying liver invasion and offer clinical value for immunotherapies.
Authors
Maolan Li, Zhaonan Liu, Shenbing Shan, Ziyao Jia, Yongsheng Li, Fatao Liu, Lina Lu, Shimei Qiu, Chen Li, Ziyi Wang, Siyuan Yan, Yuhao Zhao, Lili Gao, Zhiqing Yuan, Yuanding Liu, Jiyao Ma, Jiayi Feng, Pengxiao Geng, Yiming Li, Xiaojing Xu, Xinhua Lin, Changjun Liu, Zebing Liu, Wenguang Wu, Xiangsong Wu, Wei Gong, Yanjing Li, Dongxi Xiang, Yongning He, Yun Liu, Rong Shao, Kwan Man, Wu Wei, Yingbin Liu
Abstract
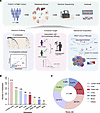
The multi-omics data represented by genomic data from patients with metastatic triple-negative breast cancer (TNBC) is crucial for precision treatment, yet data on genomic alterations in metastatic cohorts and Chinese populations remains limited. We performed targeted sequencing of 296 metastatic TNBC samples from 296 patients treated at Fudan University Shanghai Cancer Center (October 2018 to November 2020) using a 484-gene panel, identifying 796 metastatic events across 18 organ sites. We characterized the genomic landscape of TNBC metastases and identified marked enrichment of polycystin-1 (PKD1) mutations in metastatic lesions — a finding validated in an independent paired primary metastasis cohort (n = 105). Notably, PKD1 mutations were associated with resistance to anti–PD-1 therapy, as validated across 3 clinical trials (NCT03805399, NCT04129996, and NCT04395989). Multi-omics analyses, combined with functional in vitro and in vivo mechanistic studies, revealed that PKD1 modulated the “desert” tumor immune microenvironment via C-C motif chemokine ligand 2 (CCL2), and targeting CCL2 could reverse immunotherapy resistance. This comprehensive genomic characterization of metastases enhances our understanding of tumor evolution, identifies PKD1 as a previously uncharacterized regulator of immune evasion to our knowledge, and suggests a potential therapeutic strategy to overcome immunotherapy resistance.
Authors
Xiu-Zhi Zhu, Yi-Fan Zhou, Xiao-Han Ying, Yun-Yi Wang, Xiao-Hong Ding, Kun-Yu Zhang, Zhi-Ming Shao, Xi Jin, Yi-Zhou Jiang, Zhong-Hua Wang
Abstract
Single-cell analysis of human triple-negative breast cancer revealed heterogeneous macrophage populations with opposing phenotypes—pro-inflammatory and pro-resolution of inflammation. Paradoxically, both subsets accumulated in therapy-refractory residual tumors but showed inverse correlations across patients, suggesting mutually exclusive resistance mechanisms. Inflammatory macrophages localized preferentially to epithelial-like tumors, whereas pro-resolution macrophages were enriched in mesenchymal-like tumors. Mouse models faithfully recapitulated these patterns. After immuno-chemotherapy, mesenchymal-like tumors expanded pro-resolution macrophages through phagocytosis/efferocytosis, ω-3 fatty-acid uptake, and resolvin production. Macrophage-secreted C1q emerged as a principal antagonist of T-cell function by targeting mitochondria and inducing metabolic dysfunction. By contrast, epithelial-like tumors accumulated inflammatory macrophages and neutrophils that produced prostaglandins via ω-6 fatty-acid pathways. Knocking down ELOVL5—an elongase involved in ω-3 and ω-6 metabolism—mitigated both neutrophil- and macrophage-mediated immunosuppression. These distinct axes, driven by dysregulated inflammation and resolution programs, converged to undermine therapy-induced immunosurveillance; however, targeting their shared upstream regulators may overcome these resistance mechanisms.
Authors
Liqun Yu, Charlotte Rivas, Fengshuo Liu, Yichao Shen, Ling Wu, Zhan Xu, Yunfeng Ding, Xiaoxin Hao, Weijie Zhang, Hilda L. Chan, Jun Liu, Bo Wei, Yang Gao, Luis Becerra-Dominguez, Yi-Hsuan Wu, Siyue Wang, Tobie D. Lee, Xuan Li, Xiang Chen, David G. Edwards, Xiang H.-F. Zhang

Abstract
Radiation therapy (RT) is the standard of care for glioblastoma but is not curative. Triggering the cGAS/stimulator of interferon genes (STING) pathway with potent agonists, such as 8803, exerts activity across high-grade glioma preclinical models. To determine if the combination of 8803 with RT warrants consideration in the up-front treatment setting and to clarify the underlying mechanisms of therapeutic activity, C57BL/6J mice harboring intracerebral CT-2A or QPP8v gliomas were treated with RT, intratumoral 8803, or both. The treatment with the combination resulted in 80% long-term survival in the CT-2A model but not in the radiation-resistant QPP8v model. This therapeutic effect was maintained in Sting–/– CT-2A cells, highlighting the direct role of the immune system in mediating the survival benefit. Single-cell RNA-Seq identified increased nitric oxide synthase 2 (Nos2) in inflammatory tumor-associated macrophages; however, the therapeutic effect was maintained in Nos2–/– mice. Additionally, 8803 reprogrammed the blood-brain barrier (BBB) by altering the Pecam and Cd147 pathways in endothelial cells; intracranial injection of 8803 induced bihemispheric BBB opening for up to 24 hours. Sting activation was visualized longitudinally using 3’-deoxy-3’-[18F]-fluorothymidine ([18F]-FLT) PET, which peaked 72–96 hours after 8803 administration. In summary, 8803 combined with RT triggers distinctive antiglioma immune reactivity, facilitates BBB opening, and warrants consideration for up-front clinical trials in glioblastoma, where treatment effects can be monitored using [18F]-FLT PET imaging.
Authors
Shashwat Tripathi, Hinda Najem, Lisa Hurley, Ruochen Du, Crismita Dmello, Heba Ali, Kathleen McCortney, Karl J. Habashy, Peng Zhang, Craig M. Horbinski, Lara Leoni, Ryan J. Avery, Rimas V. Lukas, Timothy L. Sita, David R. Raleigh, Sean Sachdev, Roger Stupp, Maciej S. Lesniak, David M. Ashley, Daniele Procissi, Michael A. Curran, Irina Balyasnikova, Amy B. Heimberger

Abstract
The approval of sotorasib and adagrasib as the first KRAS G12C inhibitors has made the RAS oncogene a druggable target. However, they have modest objective response rates and short response durations. Therefore, strategies for improving RAS-targeted cancer therapy are urgently needed. Here, we found that both sotorasib and adagrasib promoted topoisomerase IIα (Topo IIα) proteasomal degradation in KRAS G12C–mutant cancer cells and induced DNA damage and apoptosis. In cell lines with acquired resistance to sotorasib, elevated Topo IIα levels were detected. TOP2A overexpression in sensitive KRAS G12C–mutant cells conferred resistance to sotorasib, whereas TOP2A knockdown in sotorasib-resistant cell lines sensitized the cells to sotorasib. Moreover, the combination of a KRAS G12C inhibitor such as sotorasib with a Topo II inhibitor such as VP-16 synergistically decreased the survival of sotorasib-resistant RAS G12C–mutant cells with augmented induction of DNA damage and apoptosis, effectively inhibited the growth of sotorasib-resistant tumors, and delayed or prevented the emergence of acquired resistance to sotorasib in vivo. Collectively, our results reveal an essential role of Topo IIα inhibition in mediating the therapeutic efficacy of RAS-targeted cancer therapy, providing a strong scientific rationale for targeting Topo II to improve RAS-targeted cancer therapies.
Authors
Rongzhong Xu, Dongsheng Wang, Guangzhi Ma, Xun Yuan, Qian Chu, Songqing Fan, Rener Zhang, Pan Du, Shidong Jia, Ticiana A. Leal, Suresh S. Ramalingam, Zhen Chen, Shi-Yong Sun
Abstract
Fibroblast growth factor receptor 1 (FGFR1) is recurrently mutated at p.N546 in neuroblastoma. We here sought to examine whether mutant FGFR1 is an oncogenic driver, a predictive biomarker, and an actionable vulnerability in this malignancy. FGFR1 mutations at p.N546 were associated with high-risk disease and rapid tumor progression, resulting in dismal outcome of these patients. Ectopic expression of FGFR1N546K induced constitutive down-stream signaling and interleukin-3-independent growth in Ba/F3 cells, indicating oncogene addicted proliferation. In FGFR1N546K;MYCN transgenic mice, neuroblastoma developed within the first days of life with fatal outcome within 3 weeks, reflecting the devastating clinical phenotypes of patients with FGFR1 mutant high-risk neuroblastoma. Treatment with FGFR inhibitors impaired proliferation and pathway activation in FGFR1N546K-expressing Ba/F3 and patient-derived FGFR1N546K mutant neuroblastoma cells, and inhibited tumor growth in FGFR1N546K;MYCN transgenic mice and in a chemotherapy-resistant patient-derived xenograft mouse model. In addition, partial regression of FGFR1N546K mutant tumor lesions occurred upon treatment with the FGFR inhibitor futibatinib and low-intensity chemotherapy in a patient with refractory neuroblastoma. Together, our data demonstrate that FGFR1N546K is a strong oncogenic driver in neuroblastoma that is associated with failure of current standard chemotherapy, and suggest potential clinical benefit of FGFR-directed therapies in FGFR1 mutant high-risk patients.
Authors
Lisa Werr, Jana Boland, Josephine Petersen, Fiorella Iglesias, Stefanie Höppner, Christoph Bartenhagen, Carolina Rosswog, Anna-Maria Hellmann, Yvonne Kahlert, Nadine Hemstedt, Nadliv Ibruli, Marcel A. Dammert, Boris Decarolis, Jan-Michael Werner, Florian Malchers, Kathrin Schramm, Olaf Witt, Klaus Hermann Beiske, Anne Gro Wesenberg Rognlien, Maria Winther Gunnes, Karin P.S. Langenberg, Jan Molenaar, Marie Bernkopf, Sabine Taschner-Mandl, Debbie Hughes, Sally L. George, Louis Chesler, Johannes H. Schulte, Giuseppe Barone, Mario Capasso, Lea F. Surrey, Rochelle Bagatell, Julien Masliah-Planchon, Gudrun Schleiermacher, Holger Grüll, Frank Westermann, Anne M. Schultheis, Reinhard Büttner, Anton G. Henssen, Angelika Eggert, Martin Peifer, Neerav N. Shukla, Thorsten Simon, Barbara Hero, H. Christian Reinhardt, Roman K. Thomas, Matthias Fischer
Abstract
Cervical cancer (CC) remains the fourth leading cause of cancer-related deaths in women globally, with poor prognosis for metastatic and recurrent cases. Although genomic alterations have been extensively characterized, global proteogenomic landscape of the disease is largely under-explored. Here, we present the first genome-wide proteogenomic characterization of CC, analyzing 139 tumor-normal tissue pairs using whole-genome sequencing, transcriptomics, proteomics, and phosphoproteomics. We identified four distinct molecular subtypes with unique clinical outcomes: epithelial-mesenchymal transition (EMT, C1), proliferation (C2), immune response (C3), and epithelial differentiation (C4). A four-protein classifier (CDH13, TP53BP1, NNMT, HSPB1) was developed with strong prognostic and predictive value, particularly for immunotherapy response in subtype C3. Phosphoproteomic profiling uncovered subtype-specific kinase activity, identifying actionable therapeutic targets. Our findings further revealed previously uncharacterized somatic copy number alterations, extrachromosomal DNA landscape, and human-HPV fusion peptides, with implications for genetic heterogeneity and therapeutic targets. This study enhances the understanding of cervical cancer through deeper proteogenomic insights, and facilitates the development of personalized therapeutic strategies to improve patient outcomes.
Authors
Xun Tian, Mansheng Li, Zhi Wang, Tian Fang, Yi Liu, Jin Fang, Lejing Wang, Zhichao Jiang, Xingyu Zhao, Chen Cao, Zhiqiang Yu, Meiying Yang, Songfeng Wu, Yifan Wu, Rui Tian, Hui Wang, Yunping Zhu, Zheng Hu
Abstract
Radiotherapy (RT) is a central treatment for prostate cancer (PCa), acting by inducing DNA double-strand breaks (DSBs). Tumor ability to repair these breaks limits RT efficacy, making DSB repair inhibitors potential radiosensitizers. Therefore, tumor-specific radiosensitization strategies are critically needed for PCa. Approximately 50% of PCa cases harbor the TMPRSS2-ERG gene fusion, leading to overexpression of the ERG transcription factor (ERG+). We demonstrate that ERG+ tumors shift DSB repair toward the PARP1-dependent end-joining (PARP1-EJ) pathway. Proteomic and western blot analyses revealed elevated PARP1, XRCC1, and LIG3 in ERG+ cells. PARP inhibition with olaparib increased residual γH2AX/53BP1 foci post-irradiation in ERG+ cells, indicating enhanced radiosensitization. In tissue-slice-cultures (TSCs) from 53 tumors of 40 high-risk PCa patients, olaparib selectively increased H2AX/53BP1 foci selectively in ERG+ samples. ERG+ patient-derived organoids also showed significantly delayed growth and survival when treated with olaparib plus RT compared to either treatment alone. Interestingly, ERG-negative cells within ERG+ TSCs were similarly radiosensitized by olaparib, likely through bystander effect, with residual 53BP1 foci levels comparable to ERG+ cells, confirmed by medium exchange experiments. These findings suggest that ERG expression promotes dependency on PARP1-EJ, rendering ERG+ PCa more susceptible to PARP inhibition. Combining PARP inhibitors with RT may offer a tumor-selective radiosensitization for ERG+ PCa patients.
Authors
Sabrina Köcher, Mohamed E. Elsesy, Ayham Moustafa, Wahid Mohammadi, Adriana Perugachi Heinsohn, Yamini Nagaraj, Su Jung Oh-Hohenhorst, Jan Hahn, Bente Siebels, Thomas Mair, Susanne Burdak-Rothkamm, Pierre Tennstedt, Ronald Simon, Tobias Lange, Derya Tilki, Thorsten Frenzel, Tobias Maurer, Cordula Petersen, Hartmut Schlüter, Carsten Bokemeyer, Gunhild von Amsberg, Kai Rothkamm, Wael Y. Mansour
Abstract
Chordoma are rare malignant osseous neoplasms with a striking rate of recurrence. Primary chordomas typically originate from embryonic notochord remnants, whereas recurrent chordomas usually stem from tumor cells infiltrating bone or cartilage post-surgery. Clinically, the recurrent chordomas exhibit stiffer extracellular microenvironment (ECM) than primary tumors. Intriguingly, this study identified cytoskeleton rearrangement, stress fiber reorganization, enhanced stemness, and Notch signaling activation in recurrent chordoma tissues or cell lines surviving stiff substrates, indicating the critical roles of mechanical remodeling and tumor stemness in stiffness-resistance. We propose a novel recurrence model where tumor cells experience mechanoadaptive organization to resist stiff microenvironment-induced cell death. O-GlcNAcylation of Notch1 intracellular domain (NICD1) is central to this process. Mechanistically, the stiff ECM-driven ligand-independent phosphorylation of EPHA2 sequentially activates LYN kinase, and subsequently triggers OGT activity by phosphorylating Y989 and Y418, the newly revealed critical residues for OGT glycosyltransferase activity; this induces NICD1 O-GlcNAcylation at T2063, T2090, and S2162, specifically promoting transcription of mechanical and stemness-related genes. MIR31 deletion upregulates LYN, enhancing stiffness perception and tipping the balance toward O-GlcNAc addition to NICD1, finally resulting in mechanoadaptation- and tumor stemness-driven recurrence. Consequently, MIR31 deletion is a potential biomarker for recurrence and patient stratification in Notch- or OGT-targeted therapies.
Authors
Chengjie Lian, Weiyan Peng, Peiqiang Su, Yan Ye, Jialing Liu, Dongsheng Huang, Xuejuan Sun, Yi Pu, Zhiheng Liao, Xudong Wang, Zhu Qiu, Shanshan Wu, Lei Liu







Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)