Issue published January 2, 2013 Previous issue | Next issue

- Volume 123, Issue 1
Go to section:
On the cover: Lysosomal disruption targets acute myeloid leukemia cells
Image credit: Lyndsay Stephenson.


-
Review Series
×
Abstract
Cardiovascular research is progressing on many fronts, as highlighted in the collection of Reviews in this issue of the
JCI . MicroRNAs that regulate cardiac function have been implicated in cardiac disorders, and efforts to develop therapeutic antagomirs are underway. The genetic bases of several cardiac disorders, including cardiomyopathies that cause heart failure and channelopathies that underlie cardiac arrhythmias, have been elucidated. Genetic testing can identify asymptomatic individuals at risk, potentially leading to effective preventative measures. Growing evidence supports the role of chronic inflammation in atherosclerosis, providing new opportunities for therapeutic intervention. For heart failure, recent work suggests that cardiac regeneration using stem/progenitor cells, gene transfer, new drugs that restore normal Ca2+ cycling, and agents that reduce reperfusion injury following myocardial infarction are all viable new approaches to managing disease. Cumulatively, it seems likely that the clinical advances emerging from ongoing research will, in the foreseeable future, reduce the number of deaths in the industrialized world from cardiovascular disease.Authors
Eugene Braunwald
×Abstract
The management of cardiovascular risk through lifestyle modification and pharmacotherapy is paramount to the prevention of cardiovascular disease. Epidemiological studies have identified obesity, dyslipidemia, diabetes, and hypertension as interrelated factors that negatively affect cardiovascular health. Recently, genetic and pharmacological evidence in model systems has implicated microRNAs as dynamic modifiers of disease pathogenesis. An expanded understanding of the function of microRNAs in gene regulatory networks associated with cardiovascular risk will enable identification of novel genetic mechanisms of disease and inform the development of innovative therapeutic strategies.
Authors
Daniel Quiat, Eric N. Olson
×Abstract
Genetic mutations account for a significant percentage of cardiomyopathies, which are a leading cause of congestive heart failure. In hypertrophic cardiomyopathy (HCM), cardiac output is limited by the thickened myocardium through impaired filling and outflow. Mutations in the genes encoding the thick filament components myosin heavy chain and myosin binding protein C (
MYH7 andMYBPC3 ) together explain 75% of inherited HCMs, leading to the observation that HCM is a disease of the sarcomere. Many mutations are “private” or rare variants, often unique to families. In contrast, dilated cardiomyopathy (DCM) is far more genetically heterogeneous, with mutations in genes encoding cytoskeletal, nucleoskeletal, mitochondrial, and calcium-handling proteins. DCM is characterized by enlarged ventricular dimensions and impaired systolic and diastolic function. Private mutations account for most DCMs, with few hotspots or recurring mutations. More than 50 single genes are linked to inherited DCM, including many genes that also link to HCM. Relatively few clinical clues guide the diagnosis of inherited DCM, but emerging evidence supports the use of genetic testing to identify those patients at risk for faster disease progression, congestive heart failure, and arrhythmia.Authors
Elizabeth M. McNally, Jessica R. Golbus, Megan J. Puckelwartz
×Abstract
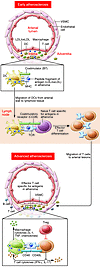
Many remarkable advances have improved our understanding of the cellular and molecular events in the pathogenesis of atherosclerosis. Chief among these is the accumulating knowledge of how the immune system contributes to all phases of atherogenesis, including well-known inflammatory reactions consequent to intimal trapping and oxidation of LDL. Advances in our understanding of the innate and adaptive responses to these events have helped to clarify the role of inflammation in atherogenesis and suggested new diagnostic modalities and novel therapeutic targets. Here we focus on recent advances in understanding how adaptive immunity affects atherogenesis.
Authors
Andrew H. Lichtman, Christoph J. Binder, Sotirios Tsimikas, Joseph L. Witztum
×Abstract
Cardiovascular disease is the number one cause of mortality in the Western world. The heart responds to many cardiopathological conditions with hypertrophic growth by enlarging individual myocytes to augment cardiac pump function and decrease ventricular wall tension. Initially, such cardiac hypertrophic growth is often compensatory, but as time progresses these changes become maladaptive. Cardiac hypertrophy is the strongest predictor for the development of heart failure, arrhythmia, and sudden death. Here we discuss therapeutic avenues emerging from molecular and genetic studies of cardiovascular disease in animal models. The majority of these are based on intracellular signaling pathways considered central to pathologic cardiac remodeling and hypertrophy, which then leads to heart failure. We focus our discussion on selected therapeutic targets that have more recently emerged and have a tangible translational potential given the available pharmacologic agents that could be readily evaluated in human clinical trials.
Authors
Jop H. van Berlo, Marjorie Maillet, Jeffery D. Molkentin
×Abstract
Ca2+-dependent signaling is highly regulated in cardiomyocytes and determines the force of cardiac muscle contraction. Ca2+ cycling refers to the release and reuptake of intracellular Ca2+ that drives muscle contraction and relaxation. In failing hearts, Ca2+ cycling is profoundly altered, resulting in impaired contractility and fatal cardiac arrhythmias. The key defects in Ca2+ cycling occur at the level of the sarcoplasmic reticulum (SR), a Ca2+ storage organelle in muscle. Defects in the regulation of Ca2+ cycling proteins including the ryanodine receptor 2, cardiac (RyR2)/Ca2+ release channel macromolecular complexes and the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase 2a (SERCA2a)/phospholamban complex contribute to heart failure. RyR2s are oxidized, nitrosylated, and PKA hyperphosphorylated, resulting in “leaky” channels in failing hearts. These leaky RyR2s contribute to depletion of Ca2+ from the SR, and the leaking Ca2+ depolarizes cardiomyocytes and triggers fatal arrhythmias. SERCA2a is downregulated and phospholamban is hypophosphorylated in failing hearts, resulting in impaired SR Ca2+ reuptake that conspires with leaky RyR2 to deplete SR Ca2+. Two new therapeutic strategies for heart failure (HF) are now being tested in clinical trials: (a) fixing the leak in RyR2 channels with a novel class of Ca2+-release channel stabilizers called Rycals and (b) increasing expression of SERCA2a to improve SR Ca2+ reuptake with viral-mediated gene therapy. There are many potential opportunities for additional mechanism-based therapeutics involving the machinery that regulates Ca2+ cycling in the heart.
Authors
Andrew R. Marks
×Abstract
Advances in understanding the molecular basis of myocardial dysfunction, together with the evolution of increasingly efficient gene transfer technology, make gene-based therapy a promising treatment option for heart conditions. Cardiovascular gene therapy has benefitted from recent advancements in vector technology, design, and delivery modalities. There is a critical need to explore new therapeutic approaches in heart failure, and gene therapy has emerged as a viable alternative. Advances in understanding of the molecular basis of myocardial dysfunction, together with the development of increasingly efficient gene transfer technology, has placed heart failure within reach of gene-based therapy. The recent successful and safe completion of a phase 2 trial targeting the cardiac sarcoplasmic/endoplasmic reticulum Ca2+ ATPase pump (SERCA2a) has the potential to open a new era for gene therapy for heart failure.
Authors
Roger J. Hajjar
×Abstract
This article discusses current understanding of myocardial biology, emphasizing the regeneration potential of the adult human heart and the mechanisms involved. In the last decade, a novel conceptual view has emerged. The heart is no longer considered a postmitotic organ, but is viewed as a self-renewing organ characterized by a resident stem cell compartment responsible for tissue homeostasis and cardiac repair following injury. Additionally, HSCs possess the ability to transdifferentiate and acquire the cardiomyocyte, vascular endothelial, and smooth muscle cell lineages. Both cardiac and hematopoietic stem cells may be used therapeutically in an attempt to reverse the devastating consequences of chronic heart failure of ischemic and nonischemic origin.
Authors
Piero Anversa, Jan Kajstura, Marcello Rota, Annarosa Leri
×Abstract
Despite many innovative advances in cardiology over the past 50 years, heart disease remains a major killer. The steady progress that continues to be made in diagnostics and therapeutics is offset by the cardiovascular consequences of the growing epidemics of obesity and diabetes. Truly innovative approaches on the horizon have been greatly influenced by new insights in cardiovascular development. In particular, research in stem cell biology, the cardiomyocyte lineage, and the interactions of the myocardium and epicardium have opened the door to new approaches for healing the injured heart.
Authors
Karl Degenhardt, Manvendra K. Singh, Jonathan A. Epstein
×Abstract
The abrupt cessation of effective cardiac function due to an aberrant heart rhythm can cause sudden and unexpected death at any age, a syndrome called sudden cardiac death (SCD). Annually, more than 300,000 cases of SCD occur in the United States alone, making this a major public health concern. Our current understanding of the mechanisms responsible for SCD has emerged from decades of basic science investigation into the normal electrophysiology of the heart, the molecular physiology of cardiac ion channels, fundamental cellular and tissue events associated with cardiac arrhythmias, and the molecular genetics of monogenic disorders of heart rhythm. This knowledge has helped shape the current diagnosis and treatment of inherited arrhythmia susceptibility syndromes associated with SCD and has provided a pathophysiological framework for understanding more complex conditions predisposing to this tragic event. This Review presents an overview of the molecular basis of SCD, with a focus on monogenic arrhythmia syndromes.
Authors
Alfred L. George Jr.
×Abstract
The discovery of the genetic basis of inherited arrhythmias has paved the way for an improved understanding of arrhythmogenesis in a wide spectrum of life-threatening conditions. In vitro expression of mutations and transgenic animal models have been instrumental in enhancing this understanding, but the applicability of results to the human heart remains unknown. The ability to differentiate induced pluripotent stem cells (iPSs) into cardiomyocytes enables the potential to generate patient-specific myocytes, which could be used to recapitulate the features of inherited arrhythmias in the context of the patient’s genetic background. Few studies have been reported on iPS-derived myocytes obtained from patients with heritable arrhythmias, but they have demonstrated the applicability of this innovative approach to the study of inherited arrhythmias. Here we review the results achieved by iPS investigations in arrhythmogenic syndromes and discuss the existing challenges to be addressed before the use of iPS-derived myocytes can become a part of personalized management of inherited arrhythmias.
Authors
Silvia G. Priori, Carlo Napolitano, Elisa Di Pasquale, Gianluigi Condorelli
×Abstract
Acute myocardial infarction (MI) is a major cause of death and disability worldwide. In patients with MI, the treatment of choice for reducing acute myocardial ischemic injury and limiting MI size is timely and effective myocardial reperfusion using either thombolytic therapy or primary percutaneous coronary intervention (PPCI). However, the process of reperfusion can itself induce cardiomyocyte death, known as myocardial reperfusion injury, for which there is still no effective therapy. A number of new therapeutic strategies currently under investigation for preventing myocardial reperfusion injury have the potential to improve clinical outcomes in patients with acute MI treated with PPCI.
Authors
Derek J. Hausenloy, Derek M. Yellon
×Abstract
Delivery of oxygen to tissues is the primary function of the cardiovascular system. NO, a gasotransmitter that signals predominantly through protein
S -nitrosylation to formS -nitrosothiols (SNOs) in target proteins, operates coordinately with oxygen in mammalian cellular systems. From this perspective, SNO-based signaling may have evolved as a major transducer of the cellular oxygen-sensing machinery that underlies global cardiovascular function. Here we review mechanisms that regulateS -nitrosylation in the context of its essential role in “systems-level” control of oxygen sensing, delivery, and utilization in the cardiovascular system, and we highlight examples of aberrantS -nitrosylation that may lead to altered oxygen homeostasis in cardiovascular diseases. Thus, through a bird’s-eye view ofS -nitrosylation in the cardiovascular system, we provide a conceptual framework that may be broadly applicable to the functioning of other cellular systems and physiological processes and that illuminates new therapeutic promise in cardiovascular medicine.Authors
Saptarsi M. Haldar, Jonathan S. Stamler











-
Commentaries
×
Abstract
In response to feeding, insulin promotes the uptake of sugar in peripheral tissues and suppresses the production of sugar, a process called gluconeogenesis, in the liver. Recent research has shown that chronic inflammation promotes insulin resistance, and in turn, chronically high glucose levels can drive inflammation. In this issue of the
JCI , Stanya et al. investigate the connection between inflammation and glucose homeostasis by analyzing the effect of the antiinflammatory cytokine IL-13. Their results suggest that IL-13 plays an unexpected role in the regulation of glucose homeostasis by modulating gluconeogenesis and may be a useful therapeutic target for treatment of diabetes and metabolic syndrome.Authors
Mariana Verdelho Machado, Yiping Yang, Anna Mae Diehl
×Abstract
Thyroid hormone is a well-known regulator of metabolic and cardiovascular functions, and signaling through thyroid receptors has differential effects on cells depending on the receptor isoform that they express. In this issue of the
JCI , Mittag et al. provide evidence that thyroid hormone receptors are essential for the formation of a population of parvalbuminergic neurons in the anterior hypothalamus, linking, for the first time, impaired thyroid hormone signaling during development to cellular deficits in the hypothalamus. Since this newly discovered cell group is predicted to play a role in regulating cardiovascular function, these findings suggest that developmental hypothyroidism may be the cause of cardiovascular disorders later in life.Authors
Douglas Forrest, Jürgen Wess

-
Research Articles
×
Abstract
The pathophysiology of the E150K mutation in the rod opsin gene associated with autosomal recessive retinitis pigmentosa (arRP) has yet to be determined. We generated knock-in mice carrying a single nucleotide change in exon 2 of the rod opsin gene resulting in the E150K mutation. This novel mouse model displayed severe retinal degeneration affecting rhodopsin’s stabilization of rod outer segments (ROS). Homozygous E150K (KK) mice exhibited early-onset retinal degeneration, with disorganized ROS structures, autofluorescent deposits in the subretinal space, and aberrant photoreceptor phagocytosis. Heterozygous (EK) mice displayed a delayed-onset milder retinal degeneration. Further, mutant receptors were mislocalized to the inner segments and perinuclear region. Though KK mouse rods displayed markedly decreased phototransduction, biochemical studies of the mutant rhodopsin revealed only minimally affected chromophore binding and G protein activation. Ablation of the chromophore by crossing KK mice with mice lacking the critical visual cycle protein LRAT slowed retinal degeneration, whereas blocking phototransduction by crossing KK mice with GNAT1-deficient mice slightly accelerated this process. This study highlights the importance of proper higher-order organization of rhodopsin in the native tissue and provides information about the signaling properties of this mutant rhodopsin. Additionally, these results suggest that patients heterozygous for the E150K mutation should be periodically reevaluated for delayed-onset retinal degeneration.
Authors
Ning Zhang, Alexander V. Kolesnikov, Beata Jastrzebska, Debarshi Mustafi, Osamu Sawada, Tadao Maeda, Christel Genoud, Andreas Engel, Vladimir J. Kefalov, Krzysztof Palczewski
×Abstract
Acute respiratory infections are responsible for more than 4 million deaths each year. Neutrophils play an essential role in the innate immune response to lung infection. These cells have an armamentarium of pattern recognition molecules and antimicrobial agents that identify and eliminate pathogens. In the setting of infection, neutrophil triggering receptor expressed on myeloid cells 1 (TREM-1) amplifies inflammatory signaling. Here we demonstrate for the first time that TREM-1 also plays an important role in transepithelial migration of neutrophils into the airspace. We developed a TREM-1/3–deficient mouse model of pneumonia and found that absence of TREM-1/3 markedly increased mortality following
Pseudomonas aeruginosa challenge. Unexpectedly, TREM-1/3 deficiency resulted in increased local and systemic cytokine production. TREM-1/3–deficient neutrophils demonstrated intact bacterial killing, phagocytosis, and chemotaxis; however, histologic examination of TREM-1/3–deficient lungs revealed decreased neutrophil infiltration of the airways. TREM-1/3–deficient neutrophils effectively migrated across primary endothelial cell monolayers but failed to migrate across primary airway epithelia grown at the air-liquid interface. These data define a new function for TREM-1 in neutrophil migration across airway epithelial cells and suggest that it amplifies inflammation through targeted neutrophil migration into the lung.Authors
Julia Klesney-Tait, Kathy Keck, Xiaopeng Li, Susan Gilfillan, Karel Otero, Sankar Baruah, David K. Meyerholz, Steven M. Varga, Cory J. Knudson, Thomas O. Moninger, Jessica Moreland, Joseph Zabner, Marco Colonna
×Abstract
Late-stage breast cancer metastasis is driven by dysregulated TGF-β signaling, but the underlying molecular mechanisms have not been fully elucidated. We attempted to recapitulate tumor and metastatic microenvironments via the use of biomechanically compliant or rigid 3D organotypic cultures and combined them with global microRNA (miR) profiling analyses to identify miRs that were upregulated in metastatic breast cancer cells by TGF-β. Here we establish miR-181a as a TGF-β–regulated “metastamir” that enhanced the metastatic potential of breast cancers by promoting epithelial-mesenchymal transition, migratory, and invasive phenotypes. Mechanistically, inactivation of miR-181a elevated the expression of the proapoptotic molecule Bim, which sensitized metastatic cells to anoikis. Along these lines, miR-181a expression was essential in driving pulmonary micrometastatic outgrowth and enhancing the lethality of late-stage mammary tumors in mice. Finally, miR-181a expression was dramatically and selectively upregulated in metastatic breast tumors, particularly triple-negative breast cancers, and was highly predictive for decreased overall survival in human breast cancer patients. Collectively, our findings strongly implicate miR-181a as a predictive biomarker for breast cancer metastasis and patient survival, and consequently, as a potential therapeutic target in metastatic breast cancer.
Authors
Molly A. Taylor, Khalid Sossey-Alaoui, Cheryl L. Thompson, David Danielpour, William P. Schiemann
×Abstract
Bacterial LPS (endotoxin) has been implicated in the pathogenesis of acute liver disease through its induction of the proinflammatory cytokine TNF-α. TNF-α is a key determinant of the outcome in a well-established mouse model of acute liver failure during septic shock. One possible mechanism for regulating TNF-α expression is through the control of protein elongation during translation, which would allow rapid cell adaptation to physiological changes. However, the regulation of translational elongation is poorly understood. We found that expression of p38γ/δ MAPK proteins is required for the elongation of nascent TNF-α protein in macrophages. The MKK3/6-p38γ/δ pathway mediated an inhibitory phosphorylation of eukaryotic elongation factor 2 (eEF2) kinase, which in turn promoted eEF2 activation (dephosphorylation) and subsequent TNF-α elongation. These results identify a new signaling pathway that regulates TNF-α production in LPS-induced liver damage and suggest potential cell-specific therapeutic targets for liver diseases in which TNF-α production is involved.
Authors
Bárbara González-Terán, José R. Cortés, Elisa Manieri, Nuria Matesanz, Ángeles Verdugo, María E. Rodríguez, Águeda González-Rodríguez, Ángela Valverde, Pilar Martín, Roger J. Davis, Guadalupe Sabio
×Abstract
TLR activation on CD11c+ DCs triggers DC maturation, which is critical for T cell activation. Given the expansion of CD11c+ DCs during the progression of atherosclerosis and the key role of T cell activation in atherogenesis, we sought to understand the role of TLR signaling in CD11c+ DCs in atherosclerosis. To this end, we used a mouse model in which a key TLR adaptor involved in DC maturation, MYD88, is deleted in CD11c+ DCs. We transplanted bone marrow containing
Myd88 -deficient CD11c+ DCs into Western diet–fed LDL receptor knockout mice and found that the transplanted mice had decreased activation of effector T cells in the periphery as well as decreased infiltration of both effector T cells and Tregs in atherosclerotic lesions. Surprisingly, the net effect was an increase in atherosclerotic lesion size due to an increase in the content of myeloid-derived inflammatory cells. The mechanism involves increased lesional monocyte recruitment associated with loss of Treg-mediated suppression of MCP-1. Thus, the dominant effect of MYD88 signaling in CD11c+ DCs in the setting of atherosclerosis is to promote the development of atheroprotective Tregs. In the absence of MYD88 signaling in CD11c+ DCs, the loss of this protective Treg response trumps the loss of proatherogenic T effector cell activation.Authors
Manikandan Subramanian, Edward Thorp, Goran K. Hansson, Ira Tabas
×Abstract
Metastasis involves critical interactions between cancer and stromal cells. Intratumoral hypoxia promotes metastasis through activation of hypoxia-inducible factors (HIFs). We demonstrate that HIFs mediate paracrine signaling between breast cancer cells (BCCs) and mesenchymal stem cells (MSCs) to promote metastasis. In a mouse orthotopic implantation model, MSCs were recruited to primary breast tumors and promoted BCC metastasis to LNs and lungs in a HIF-dependent manner. Coculture of MSCs with BCCs augmented HIF activity in BCCs. Additionally, coculture induced expression of the chemokine CXCL10 in MSCs and the cognate receptor CXCR3 in BCCs, which was augmented by hypoxia.
CXCR3 expression was blocked in cocultures treated with neutralizing antibody against CXCL10. Conversely,CXCL10 expression was blocked in MSCs cocultured with BCCs that did not express CXCR3 or HIFs. MSC coculture did not enhance the metastasis of HIF-deficient BCCs. BCCs and MSCs expressed placental growth factor (PGF) and its cognate receptor VEGFR1, respectively, in a HIF-dependent manner, and CXCL10 expression by MSCs was dependent on PGF expression by BCCs. PGF promoted metastasis of BCCs and also facilitated homing of MSCs to tumors. Thus, HIFs mediate complex and bidirectional paracrine signaling between BCCs and MSCs that stimulates breast cancer metastasis.Authors
Pallavi Chaturvedi, Daniele M. Gilkes, Carmen Chak Lui Wong, Kshitiz, Weibo Luo, Huafeng Zhang, Hong Wei, Naoharu Takano, Luana Schito, Andre Levchenko, Gregg L. Semenza
×Abstract
Influenza causes substantial morbidity and mortality, and highly pathogenic and drug-resistant strains are likely to emerge in the future. Protease-activated receptor 1 (PAR1) is a thrombin-activated receptor that contributes to inflammatory responses at mucosal surfaces. The role of PAR1 in pathogenesis of virus infections is unknown. Here, we demonstrate that PAR1 contributed to the deleterious inflammatory response after influenza virus infection in mice. Activating PAR1 by administering the agonist TFLLR-NH2 decreased survival and increased lung inflammation after influenza infection. Importantly, both administration of a PAR1 antagonist and PAR1 deficiency protected mice from infection with influenza A viruses (IAVs). Treatment with the PAR1 agonist did not alter survival of mice deficient in plasminogen (PLG), which suggests that PLG permits and/or interacts with a PAR1 function in this model. PAR1 antagonists are in human trials for other indications. Our findings suggest that PAR1 antagonism might be explored as a treatment for influenza, including that caused by highly pathogenic H5N1 and oseltamivir-resistant H1N1 viruses.
Authors
Khaled Khoufache, Fatma Berri, Wolfgang Nacken, Annette B. Vogel, Marie Delenne, Eric Camerer, Shaun R. Coughlin, Peter Carmeliet, Bruno Lina, Guus F. Rimmelzwaan, Oliver Planz, Stephan Ludwig, Béatrice Riteau
×Abstract
Brown adipose tissue (BAT) is known to function in the dissipation of chemical energy in response to cold or excess feeding, and also has the capacity to modulate energy balance. To test the hypothesis that BAT is fundamental to the regulation of glucose homeostasis, we transplanted BAT from male donor mice into the visceral cavity of age- and sex-matched recipient mice. By 8–12 weeks following transplantation, recipient mice had improved glucose tolerance, increased insulin sensitivity, lower body weight, decreased fat mass, and a complete reversal of high-fat diet–induced insulin resistance. Increasing the quantity of BAT transplanted into recipient mice further improved the metabolic effects of transplantation. BAT transplantation increased insulin-stimulated glucose uptake in vivo into endogenous BAT, white adipose tissue (WAT), and heart muscle but, surprisingly, not skeletal muscle. The improved metabolic profile was lost when the BAT used for transplantation was obtained from
Il6 –knockout mice, demonstrating that BAT-derived IL-6 is required for the profound effects of BAT transplantation on glucose homeostasis and insulin sensitivity. These findings reveal a previously under-appreciated role for BAT in glucose metabolism.Authors
Kristin I. Stanford, Roeland J.W. Middelbeek, Kristy L. Townsend, Ding An, Eva B. Nygaard, Kristen M. Hitchcox, Kathleen R. Markan, Kazuhiro Nakano, Michael F. Hirshman, Yu-Hua Tseng, Laurie J. Goodyear
×Abstract
Deposition of amyloid β protein (Aβ) to form neuritic plaques in the brain is the pathological hallmark of Alzheimer’s disease (AD). Aβ is generated from sequential cleavages of the β-amyloid precursor protein (APP) by the β- and γ-secretases, and β-site APP-cleaving enzyme 1 (BACE1) is the β-secretase essential for Aβ generation. Previous studies have indicated that glycogen synthase kinase 3 (GSK3) may play a role in APP processing by modulating γ-secretase activity, thereby facilitating Aβ production. There are two highly conserved isoforms of GSK3: GSK3α and GSK3β. We now report that specific inhibition of GSK3β, but not GSK3α, reduced BACE1-mediated cleavage of APP and Aβ production by decreasing
BACE1 gene transcription and expression. The regulation ofBACE1 gene expression by GSK3β was dependent on NF-κB signaling. Inhibition of GSK3 signaling markedly reduced Aβ deposition and neuritic plaque formation, and rescued memory deficits in the double transgenic AD model mice. These data provide evidence for regulation of BACE1 expression and AD pathogenesis by GSK3β and that inhibition of GSK3 signaling can reduce Aβ neuropathology and alleviate memory deficits in AD model mice. Our study suggests that interventions that specifically target the β-isoform of GSK3 may be a safe and effective approach for treating AD.Authors
Philip T.T. Ly, Yili Wu, Haiyan Zou, Ruitao Wang, Weihui Zhou, Ayae Kinoshita, Mingming Zhang, Yi Yang, Fang Cai, James Woodgett, Weihong Song
×Abstract
Nephrocalcinosis, acute calcium oxalate (CaOx) nephropathy, and renal stone disease can lead to inflammation and subsequent renal failure, but the underlying pathological mechanisms remain elusive. Other crystallopathies, such as gout, atherosclerosis, and asbestosis, trigger inflammation and tissue remodeling by inducing IL-1β secretion, leading us to hypothesize that CaOx crystals may induce inflammation in a similar manner. In mice, intrarenal CaOx deposition induced tubular damage, cytokine expression, neutrophil recruitment, and renal failure. We found that CaOx crystals activated murine renal DCs to secrete IL-1β through a pathway that included NLRP3, ASC, and caspase-1. Despite a similar amount of crystal deposits, intrarenal inflammation, tubular damage, and renal dysfunction were abrogated in mice deficient in MyD88; NLRP3, ASC, and caspase-1; IL-1R; or IL-18. Nephropathy was attenuated by DC depletion, ATP depletion, or therapeutic IL-1 antagonism. These data demonstrated that CaOx crystals trigger IL-1β–dependent innate immunity via the NLRP3/ASC/caspase-1 axis in intrarenal mononuclear phagocytes and directly damage tubular cells, leading to the release of the NLRP3 agonist ATP. Furthermore, these results suggest that IL-1β blockade may prevent renal damage in nephrocalcinosis.
Authors
Shrikant R. Mulay, Onkar P. Kulkarni, Khader V. Rupanagudi, Adriana Migliorini, Murthy N. Darisipudi, Akosua Vilaysane, Daniel Muruve, Yan Shi, Fay Munro, Helen Liapis, Hans-Joachim Anders
×Abstract
IL-17–producing CD8+ T (Tc17) cells are detectible in multiple sclerosis (MS) lesions; however, their contribution to the disease is unknown. To identify functions of Tc17 cells, we induced EAE, a murine model of MS, in mice lacking IFN regulatory factor 4 (IRF4). IRF4-deficient mice failed to generate Tc17 and Th17 cells and were resistant to EAE. After adoptive transfer of WT CD8+ T cells and subsequent immunization for EAE induction in these mice, the CD8+ T cells developed a Tc17 phenotype in the periphery but could not infiltrate the CNS. Similarly, transfer of small numbers of WT CD4+ T cells alone did not evoke EAE, but when transferred together with CD8+ T cells, IL-17–producing CD4+ (Th17) T cells accumulated in the CNS and mice developed severe disease. Th17 accumulation and development of EAE required IL-17A production by CD8+ T cells, suggesting that Tc17 cells are required to promote CD4+ T cell–mediated induction of EAE. Accordingly, patients with early-stage MS harbored a greater number of Tc17 cells in the cerebrospinal fluid than in peripheral blood. Our results reveal that Tc17 cells contribute to the initiation of CNS autoimmunity in mice and humans by supporting Th17 cell pathogenicity.
Authors
Magdalena Huber, Sylvia Heink, Axel Pagenstecher, Katharina Reinhard, Josephine Ritter, Alexander Visekruna, Anna Guralnik, Nadine Bollig, Katharina Jeltsch, Christina Heinemann, Eva Wittmann, Thorsten Buch, Olivia Prazeres da Costa, Anne Brüstle, Dirk Brenner, Tak W. Mak, Hans-Willi Mittrücker, Björn Tackenberg, Thomas Kamradt, Michael Lohoff
×Abstract
Hyperglycemia is a result of impaired insulin action on glucose production and disposal, and a major target of antidiabetic therapies. The study of insulin-independent regulatory mechanisms of glucose metabolism may identify new strategies to lower blood sugar levels. Here we demonstrate an unexpected metabolic function for IL-13 in the control of hepatic glucose production. IL-13 is a Th2 cytokine known to mediate macrophage alternative activation. Genetic ablation of
Il-13 in mice (Il-13–/– ) resulted in hyperglycemia, which progressed to hepatic insulin resistance and systemic metabolic dysfunction. InIl-13–/– mice, upregulation of enzymes involved in hepatic gluconeogenesis was a primary event leading to dysregulated glucose metabolism. IL-13 inhibited transcription of gluconeogenic genes by acting directly on hepatocytes through Stat3, a noncanonical downstream effector. Consequently, the ability of IL-13 to suppress glucose production was abolished in liver cells lackingStat3 or IL-13 receptor α1 (Il-13r α1 ), which suggests that the IL-13Rα1/Stat3 axis directs IL-13 signaling toward metabolic responses. These findings extend the implication of a Th1/Th2 paradigm in metabolic homeostasis beyond inflammation to direct control of glucose metabolism and suggest that the IL-13/Stat3 pathway may serve as a therapeutic target for glycemic control in insulin resistance and type 2 diabetes.Authors
Kristopher J. Stanya, David Jacobi, Sihao Liu, Prerna Bhargava, Lingling Dai, Matthew R. Gangl, Karen Inouye, Jillian L. Barlow, Yewei Ji, Joseph P. Mizgerd, Ling Qi, Hang Shi, Andrew N.J. McKenzie, Chih-Hao Lee
×Abstract
A cell-based therapy for the replacement of dopaminergic neurons has been a long-term goal in Parkinson’s disease research. Here, we show that autologous engraftment of A9 dopaminergic neuron-like cells induced from mesenchymal stem cells (MSCs) leads to long-term survival of the cells and restoration of motor function in hemiparkinsonian macaques. Differentiated MSCs expressed markers of A9 dopaminergic neurons and released dopamine after depolarization in vitro. The differentiated autologous cells were engrafted in the affected portion of the striatum. Animals that received transplants showed modest and gradual improvements in motor behaviors. Positron emission tomography (PET) using [11C]-CFT, a ligand for the dopamine transporter (DAT), revealed a dramatic increase in DAT expression, with a subsequent exponential decline over a period of 7 months. Kinetic analysis of the PET findings revealed that DAT expression remained above baseline levels for over 7 months. Immunohistochemical evaluations at 9 months consistently demonstrated the existence of cells positive for DAT and other A9 dopaminergic neuron markers in the engrafted striatum. These data suggest that transplantation of differentiated autologous MSCs may represent a safe and effective cell therapy for Parkinson’s disease.
Authors
Takuya Hayashi, Shohei Wakao, Masaaki Kitada, Takayuki Ose, Hiroshi Watabe, Yasumasa Kuroda, Kanae Mitsunaga, Dai Matsuse, Taeko Shigemoto, Akihito Ito, Hironobu Ikeda, Hidenao Fukuyama, Hirotaka Onoe, Yasuhiko Tabata, Mari Dezawa
×Abstract
MicroRNAs (miRNAs) and methionine adenosyltransferase 1A (
MAT1A ) are dysregulated in hepatocellular carcinoma (HCC), and reducedMAT1A expression correlates with worse HCC prognosis. Expression of miR-664, miR-485-3p, and miR-495, potential regulatory miRNAs ofMAT1A , is increased in HCC. Knockdown of these miRNAs individually in Hep3B and HepG2 cells inducedMAT1A expression, reduced growth, and increased apoptosis, while combined knockdown exerted additional effects on all parameters. Subcutaneous and intraparenchymal injection of Hep3B cells stably overexpressing each of this trio of miRNAs promoted tumorigenesis and metastasis in mice. Treatment with miRNA-664 (miR-664), miR-485-3p, and miR-495 siRNAs reduced tumor growth, invasion, and metastasis in an orthotopic liver cancer model. BlockingMAT1A induction significantly reduced the antitumorigenic effect of miR-495 siRNA, whereas maintainingMAT1A expression prevented miRNA-mediated enhancement of growth and metastasis. Knockdown of these miRNAs increased total and nuclear level of MAT1A protein, global CpG methylation, lin-28 homolog B (Caenorhabditis elegans ) (LIN28B ) promoter methylation, and reducedLIN28B expression. The opposite occurred with forced expression of these miRNAs. In conclusion, upregulation of miR-664, miR-485-3p, and miR-495 contributes to lowerMAT1A expression in HCC, and enhanced tumorigenesis may provide potential targets for HCC therapy.Authors
Heping Yang, Michele E. Cho, Tony W.H. Li, Hui Peng, Kwang Suk Ko, Jose M. Mato, Shelly C. Lu
×Abstract
Aberrant expression of the homeodomain transcription factor CDX2 occurs in most cases of acute myeloid leukemia (AML) and promotes leukemogenesis, making CDX2, in principle, an attractive therapeutic target. Conversely, CDX2 acts as a tumor suppressor in colonic epithelium. The effectors mediating the leukemogenic activity of CDX2 and the mechanism underlying its context-dependent properties are poorly characterized, and strategies for interfering with CDX2 function in AML remain elusive. We report data implicating repression of the transcription factor KLF4 as important for the oncogenic activity of CDX2, and demonstrate that CDX2 differentially regulates KLF4 in AML versus colon cancer cells through a mechanism that involves tissue-specific patterns of promoter binding and epigenetic modifications. Furthermore, we identified deregulation of the PPARγ signaling pathway as a feature of CDX2-associated AML and observed that PPARγ agonists derepressed KLF4 and were preferentially toxic to CDX2+ leukemic cells. These data delineate transcriptional programs associated with CDX2 expression in hematopoietic cells, provide insight into the antagonistic duality of CDX2 function in AML versus colon cancer, and suggest reactivation of KLF4 expression, through modulation of PPARγ signaling, as a therapeutic modality in a large proportion of AML patients.
Authors
Katrin Faber, Lars Bullinger, Christine Ragu, Angela Garding, Daniel Mertens, Christina Miller, Daniela Martin, Daniel Walcher, Konstanze Döhner, Hartmut Döhner, Rainer Claus, Christoph Plass, Stephen M. Sykes, Steven W. Lane, Claudia Scholl, Stefan Fröhling
×Abstract
Despite efforts to understand and treat acute myeloid leukemia (AML), there remains a need for more comprehensive therapies to prevent AML-associated relapses. To identify new therapeutic strategies for AML, we screened a library of on- and off-patent drugs and identified the antimalarial agent mefloquine as a compound that selectively kills AML cells and AML stem cells in a panel of leukemia cell lines and in mice. Using a yeast genome-wide functional screen for mefloquine sensitizers, we identified genes associated with the yeast vacuole, the homolog of the mammalian lysosome. Consistent with this, we determined that mefloquine disrupts lysosomes, directly permeabilizes the lysosome membrane, and releases cathepsins into the cytosol. Knockdown of the lysosomal membrane proteins LAMP1 and LAMP2 resulted in decreased cell viability, as did treatment of AML cells with known lysosome disrupters. Highlighting a potential therapeutic rationale for this strategy, leukemic cells had significantly larger lysosomes compared with normal cells, and leukemia-initiating cells overexpressed lysosomal biogenesis genes. These results demonstrate that lysosomal disruption preferentially targets AML cells and AML progenitor cells, providing a rationale for testing lysosomal disruption as a novel therapeutic strategy for AML.
Authors
Mahadeo A. Sukhai, Swayam Prabha, Rose Hurren, Angela C. Rutledge, Anna Y. Lee, Shrivani Sriskanthadevan, Hong Sun, Xiaoming Wang, Marko Skrtic, Ayesh Seneviratne, Maria Cusimano, Bozhena Jhas, Marcela Gronda, Neil MacLean, Eunice E. Cho, Paul A. Spagnuolo, Sumaiya Sharmeen, Marinella Gebbia, Malene Urbanus, Kolja Eppert, Dilan Dissanayake, Alexia Jonet, Alexandra Dassonville-Klimpt, Xiaoming Li, Alessandro Datti, Pamela S. Ohashi, Jeff Wrana, Ian Rogers, Pascal Sonnet, William Y. Ellis, Seth J. Corey, Connie Eaves, Mark D. Minden, Jean C.Y. Wang, John E. Dick, Corey Nislow, Guri Giaever, Aaron D. Schimmer
×Abstract
Neurofibromatosis type 1 (NF1) predisposes individuals to the development of juvenile myelomonocytic leukemia (JMML), a fatal myeloproliferative disease (MPD). In genetically engineered murine models, nullizygosity of
Nf1 , a tumor suppressor gene that encodes a Ras-GTPase–activating protein, results in hyperactivity of Raf/Mek/Erk in hematopoietic stem and progenitor cells (HSPCs). Activated Erk1/2 phosphorylate kinases and transcription factors with myriad mitogenic roles in diverse cell types. However, genetic studies examining Erk1/2’s differential and/or combined control of normal andNf1 -deficient myelopoiesis are lacking. Moreover, prior studies relying on chemical Mek/Erk inhibitors have reached conflicting conclusions in normal andNf1 -deficient mice. Here, we show that while singleErk1 orErk2 disruption did not grossly compromise myelopoiesis, dualErk1/2 disruption rapidly ablated granulocyte and monocyte production in vivo, diminished progenitor cell number, and prevented HSPC proliferation in vitro. Genetic disruption ofErk1/2 in the context ofNf1 nullizygosity (Mx1Cre+Nf1flox/floxErk1–/–Erk2flox/flox ) fully protects against the development of MPD. Collectively, we identified a fundamental requirement for Erk1/2 signaling in normal andNf1 -deficient hematopoiesis, elucidating a critical hematopoietic function for Erk1/2 while genetically validating highly selective Mek/Erk inhibitors in a leukemia that is otherwise resistant to traditional therapy.Authors
Karl Staser, Su-Jung Park, Steven D. Rhodes, Yi Zeng, Yong Zheng He, Matthew A. Shew, Jeffrey R. Gehlhausen, Donna Cerabona, Keshav Menon, Shi Chen, Zejin Sun, Jin Yuan, David A. Ingram, Grzegorz Nalepa, Feng-Chun Yang, D. Wade Clapp
×Abstract
Children with neurofibromatosis type 1 (NF1) are predisposed to juvenile myelomonocytic leukemia (JMML), an aggressive myeloproliferative neoplasm (MPN) that is refractory to conventional chemotherapy. Conditional inactivation of the
Nf1 tumor suppressor in hematopoietic cells of mice causes a progressive MPN that accurately models JMML and chronic myelomonocytic leukemia (CMML). We characterized the effects ofNf1 loss on immature hematopoietic populations and investigated treatment with the MEK inhibitor PD0325901 (hereafter called 901). SomaticNf1 inactivation resulted in a marked expansion of immature and lineage-committed myelo-erythroid progenitors and ineffective erythropoiesis. Treatment with 901 induced a durable drop in leukocyte counts, enhanced erythropoietic function, and markedly reduced spleen sizes in mice with MPN. MEK inhibition also restored a normal pattern of erythroid differentiation and greatly reduced extramedullary hematopoiesis. Remarkably, genetic analysis revealed the persistence ofNf1 -deficient hematopoietic cells, indicating that MEK inhibition modulates the proliferation and differentiation ofNf1 mutant cells in vivo rather than eliminating them. These data provide a rationale for performing clinical trials of MEK inhibitors in patients with JMML and CMML.Authors
Tiffany Chang, Kimberly Krisman, Emily Harding Theobald, Jin Xu, Jon Akutagawa, Jennifer O. Lauchle, Scott Kogan, Benjamin S. Braun, Kevin Shannon
×Abstract
Neurofibromatosis type 1 (NF1) patients develop benign neurofibromas and malignant peripheral nerve sheath tumors (MPNST). These incurable peripheral nerve tumors result from loss of
NF1 tumor suppressor gene function, causing hyperactive Ras signaling. Activated Ras controls numerous downstream effectors, but specific pathways mediating the effects of hyperactive Ras in NF1 tumors are unknown. We performed cross-species transcriptome analyses of mouse and human neurofibromas and MPNSTs and identified global negative feedback of genes that regulate Ras/Raf/MEK/ERK signaling in both species. Nonetheless, ERK activation was sustained in mouse and human neurofibromas and MPNST. We used a highly selective pharmacological inhibitor of MEK, PD0325901, to test whether sustained Ras/Raf/MEK/ERK signaling contributes to neurofibroma growth in a neurofibromatosis mouse model (Nf1fl/fl;Dhh-Cre ) or in NF1 patient MPNST cell xenografts. PD0325901 treatment reduced aberrantly proliferating cells in neurofibroma and MPNST, prolonged survival of mice implanted with human MPNST cells, and shrank neurofibromas in more than 80% of mice tested. Our data demonstrate that deregulated Ras/ERK signaling is critical for the growth of NF1 peripheral nerve tumors and provide a strong rationale for testing MEK inhibitors in NF1 clinical trials.Authors
Walter J. Jessen, Shyra J. Miller, Edwin Jousma, Jianqiang Wu, Tilat A. Rizvi, Meghan E. Brundage, David Eaves, Brigitte Widemann, Mi-Ok Kim, Eva Dombi, Jessica Sabo, Atira Hardiman Dudley, Michiko Niwa-Kawakita, Grier P. Page, Marco Giovannini, Bruce J. Aronow, Timothy P. Cripe, Nancy Ratner
×Abstract
Down syndrome (DS) patients exhibit abnormalities of hippocampal-dependent explicit memory, a feature that is replicated in relevant mouse models of the disease. Adult hippocampal neurogenesis, which is impaired in DS and other neuropsychiatric diseases, plays a key role in hippocampal circuit plasticity and has been implicated in learning and memory. However, it remains unknown whether increasing adult neurogenesis improves hippocampal plasticity and behavioral performance in the multifactorial context of DS. We report that, in the Ts65Dn mouse model of DS, chronic administration of lithium, a clinically used mood stabilizer, promoted the proliferation of neuronal precursor cells through the pharmacological activation of the Wnt/β-catenin pathway and restored adult neurogenesis in the hippocampal dentate gyrus (DG) to physiological levels. The restoration of adult neurogenesis completely rescued the synaptic plasticity of newborn neurons in the DG and led to the full recovery of behavioral performance in fear conditioning, object location, and novel object recognition tests. These findings indicate that reestablishing a functional population of hippocampal newborn neurons in adult DS mice rescues hippocampal plasticity and memory and implicate adult neurogenesis as a promising therapeutic target to alleviate cognitive deficits in DS patients.
Authors
Andrea Contestabile, Barbara Greco, Diego Ghezzi, Valter Tucci, Fabio Benfenati, Laura Gasparini
×Abstract
Low-grade chronic inflammation is a major characteristic of obesity and results from deregulated white adipose tissue function. Consequently, there is interest in identifying the underlying regulatory mechanisms and components that drive adipocyte inflammation. Here, we report that expression of the transcriptional corepressor complex subunits GPS2 and SMRT was significantly reduced in obese adipose tissue, inversely correlated to inflammatory status, and was restored upon gastric bypass surgery–induced weight loss in morbid obesity. These alterations correlated with reduced occupancy of the corepressor complex at inflammatory promoters, providing a mechanistic explanation for elevated inflammatory transcription. In support of these correlations, RNAi-mediated depletion of GPS2 and SMRT from cultured human adipocytes promoted derepression of inflammatory transcription and elevation of obesity-associated inflammatory markers, such as IL-6 and MCP-1. Furthermore, we identified a regulatory cascade containing PPARγ and TWIST1 that controlled the expression of GPS2 and SMRT in human adipocytes. These findings were clinically relevant, because treatment of diabetic obese patients with pioglitazone, an antidiabetic and antiinflammatory PPARγ agonist, restored expression of TWIST1, GPS2, and SMRT in adipose tissue. Collectively, our findings identify alterations in a regulatory transcriptional network in adipocytes involving the dysregulation of a specific corepressor complex as among the initiating events promoting adipose tissue inflammation in human obesity.
Authors
Amine Toubal, Karine Clément, Rongrong Fan, Patricia Ancel, Veronique Pelloux, Christine Rouault, Nicolas Veyrie, Agnes Hartemann, Eckardt Treuter, Nicolas Venteclef
×Abstract
HIV-1 accumulates mutations in and around reactive epitopes to escape recognition and killing by CD8+ T cells. Measurements of HIV-1 time to escape should therefore provide information on which parameters are most important for T cell–mediated in vivo control of HIV-1. Primary HIV-1–specific T cell responses were fully mapped in 17 individuals, and the time to virus escape, which ranged from days to years, was measured for each epitope. While higher magnitude of an individual T cell response was associated with more rapid escape, the most significant T cell measure was its relative immunodominance measured in acute infection. This identified subject-level or “vertical” immunodominance as the primary determinant of in vivo CD8+ T cell pressure in HIV-1 infection. Conversely, escape was slowed significantly by lower population variability, or entropy, of the epitope targeted. Immunodominance and epitope entropy combined to explain half of all the variability in time to escape. These data explain how CD8+ T cells can exert significant and sustained HIV-1 pressure even when escape is very slow and that within an individual, the impacts of other T cell factors on HIV-1 escape should be considered in the context of immunodominance.
Authors
Michael K.P. Liu, Natalie Hawkins, Adam J. Ritchie, Vitaly V. Ganusov, Victoria Whale, Simon Brackenridge, Hui Li, Jeffrey W. Pavlicek, Fangping Cai, Melissa Rose-Abrahams, Florette Treurnicht, Peter Hraber, Catherine Riou, Clive Gray, Guido Ferrari, Rachel Tanner, Li-Hua Ping, Jeffrey A. Anderson, Ronald Swanstrom, Myron Cohen, Salim S. Abdool Karim, Barton Haynes, Persephone Borrow, Alan S. Perelson, George M. Shaw, Beatrice H. Hahn, Carolyn Williamson, Bette T. Korber, Feng Gao, Steve Self, Andrew McMichael, Nilu Goonetilleke
×Abstract
The detection of estrogen receptor-α (ERα) in osteoblasts and osteoclasts over 20 years ago suggested that direct effects of estrogens on both of these cell types are responsible for their beneficial effects on the skeleton, but the role of ERα in osteoblast lineage cells has remained elusive. In addition, estrogen activation of ERα in osteoclasts can only account for the protective effect of estrogens on the cancellous, but not the cortical, bone compartment that represents 80% of the entire skeleton. Here, we deleted
ER α at different stages of differentiation in murine osteoblast lineage cells. We found that ERα in osteoblast progenitors expressing Osterix1 (Osx1) potentiates Wnt/β-catenin signaling, thereby increasing proliferation and differentiation of periosteal cells. Further, this signaling pathway was required for optimal cortical bone accrual at the periosteum in mice. Notably, this function did not require estrogens. The osteoblast progenitorER α mediated a protective effect of estrogens against endocortical, but not cancellous, bone resorption. ERα in mature osteoblasts or osteocytes did not influence cancellous or cortical bone mass. Hence, the ERα in both osteoblast progenitors and osteoclasts functions to optimize bone mass but at distinct bone compartments and in response to different cues.Authors
Maria Almeida, Srividhya Iyer, Marta Martin-Millan, Shoshana M. Bartell, Li Han, Elena Ambrogini, Melda Onal, Jinhu Xiong, Robert S. Weinstein, Robert L. Jilka, Charles A. O’Brien, Stavros C. Manolagas
×Abstract
High-grade gliomas (HGGs) are incurable brain tumors that are characterized by the presence of glioma-initiating cells (GICs). GICs are essential to tumor aggressiveness and retain the capacity for self-renewal and multilineage differentiation as long as they reside in the perivascular niche. ID proteins are master regulators of stemness and anchorage to the extracellular niche microenvironment, suggesting that they may play a role in maintaining GICs. Here, we modeled the probable therapeutic impact of ID inactivation in HGG by selective ablation of
Id in tumor cells and after tumor initiation in a new mouse model of human mesenchymal HGG. Deletion of 3Id genes induced rapid release of GICs from the perivascular niche, followed by tumor regression. GIC displacement was mediated by derepression ofRap1gap and subsequent inhibition of RAP1, a master regulator of cell adhesion. We identified a signature module of 5 genes in the ID pathway, includingRAP1GAP , which segregated 2 subgroups of glioma patients with markedly different clinical outcomes. The model-informed survival analysis together with genetic and functional studies establish that ID activity is required for the maintenance of mesenchymal HGG and suggest that pharmacological inactivation of ID proteins could serve as a therapeutic strategy.Authors
Francesco Niola, Xudong Zhao, Devendra Singh, Ryan Sullivan, Angelica Castano, Antonio Verrico, Pietro Zoppoli, Dinorah Friedmann-Morvinski, Erik Sulman, Lindy Barrett, Yuan Zhuang, Inder Verma, Robert Benezra, Ken Aldape, Antonio Iavarone, Anna Lasorella
×Abstract
Little is known about the transcriptional regulation of tumor angiogenesis, and tumor ECs (tECs) remain poorly characterized. Here, we studied the expression pattern of the transcription factor
Sox17 in the vasculature of murine and human tumors and investigated the function ofSox17 during tumor angiogenesis usingSox17 genetic mouse models.Sox17 was specifically expressed in tECs in a heterogeneous pattern; in particular, strongSox17 expression distinguished tECs with high VEGFR2 expression. Whereas overexpression ofSox17 in tECs promoted tumor angiogenesis and vascular abnormalities,Sox17 deletion in tECs reduced tumor angiogenesis and normalized tumor vessels, inhibiting tumor growth. Tumor vessel normalization bySox17 deletion was long lasting, improved anticancer drug delivery into tumors, and inhibited tumor metastasis. Sox17 promoted endothelial sprouting behavior and upregulated VEGFR2 expression in a cell-intrinsic manner. Moreover, Sox17 increased the percentage of tumor-associated CD11b+Gr-1+ myeloid cells within tumors. The vascular effects of Sox17 persisted throughout tumor growth. Interestingly, Sox17 expression specific to tECs was also observed in highly vascularized human glioblastoma samples. Our findings establish Sox17 as a key regulator of tumor angiogenesis and tumor progression.Authors
Hanseul Yang, Sungsu Lee, Seungjoo Lee, Kangsan Kim, Yeseul Yang, Jeong Hoon Kim, Ralf H. Adams, James M. Wells, Sean J. Morrison, Gou Young Koh, Injune Kim
×Abstract
Primary immune thrombocytopenia (ITP) is a disorder caused by autoantibody-mediated platelet destruction and decreased platelet production. Rituximab, a B cell–depleting agent, has become the first-line treatment for ITP; however, patients with refractory disease usually require splenectomy. We identified antibody-secreting cells as the major splenic B cell population that is resistant to rituximab. The phenotype, antibody specificity, and gene expression profile of these cells were characterized and compared to those of antibody-secreting cells from untreated ITP spleens and from healthy tissues. Antiplatelet-specific plasma cells (PC) were detected in the spleens of patients with ITP up to 6 months after rituximab treatment, and the PC population displayed a long-lived program similar to the one of bone marrow PC, thus explaining for most of these patients the absence of response to rituximab and the response to splenectomy. When analyzed by multiplex PCR at the single-cell level, normal splenic PC showed a markedly different gene expression profile, with an intermediate signature, including genes characteristic of both long-lived PC and proliferating plasmablasts. Surprisingly, long-lived PC were not detected in untreated ITP spleens. These results suggest that the milieu generated by B cell depletion promotes the differentiation and settlement of long-lived PC in the spleen.
Authors
Matthieu Mahévas, Pauline Patin, François Huetz, Marc Descatoire, Nicolas Cagnard, Christine Bole-Feysot, Simon Le Gallou, Mehdi Khellaf, Olivier Fain, David Boutboul, Lionel Galicier, Mikael Ebbo, Olivier Lambotte, Mohamed Hamidou, Philippe Bierling, Bertrand Godeau, Marc Michel, Jean-Claude Weill, Claude-Agnès Reynaud
×Abstract
N -formyl peptide receptors (FPRs) are critical regulators of host defense in phagocytes and are also expressed in epithelia. FPR signaling and function have been extensively studied in phagocytes, yet their functional biology in epithelia is poorly understood. We describe a novel intestinal epithelial FPR signaling pathway that is activated by an endogenous FPR ligand, annexin A1 (ANXA1), and its cleavage product Ac2-26, which mediate activation of ROS by an epithelial NADPH oxidase, NOX1. We show that epithelial cell migration was regulated by this signaling cascade through oxidative inactivation of the regulatory phosphatases PTEN and PTP-PEST, with consequent activation of focal adhesion kinase (FAK) and paxillin. In vivo studies using intestinal epithelial specificNox1–/–IEC andAnxA1–/– mice demonstrated defects in intestinal mucosal wound repair, while systemic administration of ANXA1 promoted wound recovery in a NOX1-dependent fashion. Additionally, increased ANXA1 expression was observed in the intestinal epithelium and infiltrating leukocytes in the mucosa of ulcerative colitis patients compared with normal intestinal mucosa. Our findings delineate a novel epithelial FPR1/NOX1-dependent redox signaling pathway that promotes mucosal wound repair.Authors
Giovanna Leoni, Ashfaqul Alam, Philipp-Alexander Neumann, J. David Lambeth, Guangjie Cheng, James McCoy, Roland S. Hilgarth, Kousik Kundu, Niren Murthy, Dennis Kusters, Chris Reutelingsperger, Mauro Perretti, Charles A. Parkos, Andrew S. Neish, Asma Nusrat
×Abstract
Postprandially, the liver experiences an extensive metabolic reprogramming that is required for the switch from glucose production to glucose assimilation. Upon refeeding, the unfolded protein response (UPR) is rapidly, though only transiently, activated. Activation of the UPR results in a cessation of protein translation, increased chaperone expression, and increased ER-mediated protein degradation, but it is not clear how the UPR is involved in the postprandial switch to alternate fuel sources. Activation of the inositol-requiring enzyme 1 (IRE1) branch of the UPR signaling pathway triggers expression of the transcription factor Xbp1s. Using a mouse model with liver-specific inducible Xbp1s expression, we demonstrate that Xbp1s is sufficient to provoke a metabolic switch characteristic of the postprandial state, even in the absence of caloric influx. Mechanistically, we identified UDP-galactose-4-epimerase (GalE) as a direct transcriptional target of Xbp1s and as the key mediator of this effect. Our results provide evidence that the Xbp1s/GalE pathway functions as a novel regulatory nexus connecting the UPR to the characteristic postprandial metabolic changes in hepatocytes.
Authors
Yingfeng Deng, Zhao V. Wang, Caroline Tao, Ningguo Gao, William L. Holland, Anwarul Ferdous, Joyce J. Repa, Guosheng Liang, Jin Ye, Mark A. Lehrman, Joseph A. Hill, Jay D. Horton, Philipp E. Scherer
×Abstract
The scaffold protein p62 (sequestosome 1; SQSTM1) is an emerging key molecular link among the metabolic, immune, and proliferative processes of the cell. Here, we report that adipocyte-specific, but not CNS-, liver-, muscle-, or myeloid-specific
p62 -deficient mice are obese and exhibit a decreased metabolic rate caused by impaired nonshivering thermogenesis. Our results show that p62 regulates energy metabolism via control of mitochondrial function in brown adipose tissue (BAT). Accordingly, adipocyte-specificp62 deficiency led to impaired mitochondrial function, causing BAT to become unresponsive to β-adrenergic stimuli. Ablation of p62 leads to decreased activation of p38 targets, affecting signaling molecules that control mitochondrial function, such as ATF2, CREB, PGC1α, DIO2, NRF1, CYTC, COX2, ATP5β, and UCP1.p62 ablation in HIB1B and BAT primary cells demonstrated that p62 controls thermogenesis in a cell-autonomous manner, independently of brown adipocyte development or differentiation. Together, our data identify p62 as a novel regulator of mitochondrial function and brown fat thermogenesis.Authors
Timo D. Müller, Sang Jun Lee, Martin Jastroch, Dhiraj Kabra, Kerstin Stemmer, Michaela Aichler, Bill Abplanalp, Gayathri Ananthakrishnan, Nakul Bhardwaj, Sheila Collins, Senad Divanovic, Max Endele, Brian Finan, Yuanqing Gao, Kirk M. Habegger, Jazzmin Hembree, Kristy M. Heppner, Susanna Hofmann, Jenna Holland, Daniela Küchler, Maria Kutschke, Radha Krishna, Maarit Lehti, Rebecca Oelkrug, Nickki Ottaway, Diego Perez-Tilve, Christine Raver, Axel K. Walch, Sonja C. Schriever, John Speakman, Yu-Hua Tseng, Maria Diaz-Meco, Paul T. Pfluger, Jorge Moscat, Matthias H. Tschöp
×Abstract
Gastric adenocarcinoma is strongly associated with
Helicobacter pylori infection; however, most infected persons never develop this malignancy.H. pylori strains harboring thecag pathogenicity island (cag+ ), which encodes CagA and a type IV secretion system (T4SS), induce more severe disease outcomes.H. pylori infection is also associated with iron deficiency, which similarly augments gastric cancer risk. To define the influence of iron deficiency on microbial virulence in gastric carcinogenesis, Mongolian gerbils were maintained on iron-depleted diets and infected with an oncogenicH. pylori cag+ strain. Iron depletion accelerated the development ofH. pylori –induced premalignant and malignant lesions in acagA -dependent manner.H. pylori strains harvested from iron-depleted gerbils or grown under iron-limiting conditions exhibited enhanced virulence and induction of inflammatory factors. Further, in a human population at high risk for gastric cancer,H. pylori strains isolated from patients with the lowest ferritin levels induced more robust proinflammatory responses compared with strains isolated from patients with the highest ferritin levels, irrespective of histologic status. These data demonstrate that iron deficiency enhancesH. pylori virulence and represents a measurable biomarker to identify populations of infected persons at high risk for gastric cancer.Authors
Jennifer M. Noto, Jennifer A. Gaddy, Josephine Y. Lee, M. Blanca Piazuelo, David B. Friedman, Daniel C. Colvin, Judith Romero-Gallo, Giovanni Suarez, John Loh, James C. Slaughter, Shumin Tan, Douglas R. Morgan, Keith T. Wilson, Luis E. Bravo, Pelayo Correa, Timothy L. Cover, Manuel R. Amieva, Richard M. Peek Jr.
×Abstract
Cyclin D1b is a splice variant of the cell cycle regulator cyclin D1 and is known to harbor divergent and highly oncogenic functions in human cancer. While cyclin D1b is induced during disease progression in many cancer types, the mechanisms underlying cyclin D1b function remain poorly understood. Herein, cell and human tumor xenograft models of prostate cancer were utilized to resolve the downstream pathways that are required for the protumorigenic functions of cyclin D1b. Specifically, cyclin D1b was found to modulate the expression of a large transcriptional network that cooperates with androgen receptor (AR) signaling to enhance tumor cell growth and invasive potential. Notably, cyclin D1b promoted AR-dependent activation of genes associated with metastatic phenotypes. Further exploration determined that transcriptional induction of
SNAI2 (Slug) was essential for cyclin D1b–mediated proliferative and invasive properties, implicating Slug as a critical driver of disease progression. Importantly, cyclin D1b expression highly correlated with that of Slug in clinical samples of advanced disease. In vivo analyses provided strong evidence that Slug enhances both tumor growth and metastatic phenotypes. Collectively, these findings reveal the underpinning mechanisms behind the protumorigenic functions of cyclin D1b and demonstrate that the convergence of the cyclin D1b/AR and Slug pathways results in the activation of processes critical for the promotion of lethal tumor phenotypes.Authors
Michael A. Augello, Craig J. Burd, Ruth Birbe, Christopher McNair, Adam Ertel, Michael S. Magee, Daniel E. Frigo, Kari Wilder-Romans, Mark Shilkrut, Sumin Han, Danielle L. Jernigan, Jeffry L. Dean, Alessandro Fatatis, Donald P. McDonnell, Tapio Visakorpi, Felix Y. Feng, Karen E. Knudsen
×Abstract
Thyroid hormone is well known for its profound direct effects on cardiovascular function and metabolism. Recent evidence, however, suggests that the hormone also regulates these systems indirectly through the central nervous system. While some of the molecular mechanisms underlying the hormone’s central control of metabolism have been identified, its actions in the central cardiovascular control have remained enigmatic. Here, we describe a previously unknown population of parvalbuminergic neurons in the anterior hypothalamus that requires thyroid hormone receptor signaling for proper development. Specific stereotaxic ablation of these cells in the mouse resulted in hypertension and temperature-dependent tachycardia, indicating a role in the central autonomic control of blood pressure and heart rate. Moreover, the neurons exhibited intrinsic temperature sensitivity in patch-clamping experiments, providing a new connection between cardiovascular function and core temperature. Thus, the data identify what we believe to be a novel hypothalamic cell population potentially important for understanding hypertension and indicate developmental hypothyroidism as an epigenetic risk factor for cardiovascular disorders. Furthermore, the findings may be beneficial for treatment of the recently identified patients that have a mutation in thyroid hormone receptor α1.
Authors
Jens Mittag, David J. Lyons, Johan Sällström, Milica Vujovic, Susi Dudazy-Gralla, Amy Warner, Karin Wallis, Anneke Alkemade, Kristina Nordström, Hannah Monyer, Christian Broberger, Anders Arner, Björn Vennström
×Abstract
Because of the high risk of recurrence in high-grade serous ovarian carcinoma (HGS-OvCa), the development of outcome predictors could be valuable for patient stratification. Using the catalog of The Cancer Genome Atlas (TCGA), we developed subtype and survival gene expression signatures, which, when combined, provide a prognostic model of HGS-OvCa classification, named “
Cl assification ofOvar ian Cancer” (CLOVAR). We validated CLOVAR on an independent dataset consisting of 879 HGS-OvCa expression profiles. The worst outcome group, accounting for 23% of all cases, was associated with a median survival of 23 months and a platinum resistance rate of 63%, versus a median survival of 46 months and platinum resistance rate of 23% in other cases. Associating the outcome prediction model withBRCA1 /BRCA2 mutation status, residual disease after surgery, and disease stage further optimized outcome classification. Ovarian cancer is a disease in urgent need of more effective therapies. The spectrum of outcomes observed here and their association with CLOVAR signatures suggests variations in underlying tumor biology. Prospective validation of the CLOVAR model in the context of additional prognostic variables may provide a rationale for optimal combination of patient and treatment regimens.Authors
Roel G.W. Verhaak, Pablo Tamayo, Ji-Yeon Yang, Diana Hubbard, Hailei Zhang, Chad J. Creighton, Sian Fereday, Michael Lawrence, Scott L. Carter, Craig H. Mermel, Aleksandar D. Kostic, Dariush Etemadmoghadam, Gordon Saksena, Kristian Cibulskis, Sekhar Duraisamy, Keren Levanon, Carrie Sougnez, Aviad Tsherniak, Sebastian Gomez, Robert Onofrio, Stacey Gabriel, Lynda Chin, Nianxiang Zhang, Paul T. Spellman, Yiqun Zhang, Rehan Akbani, Katherine A. Hoadley, Ari Kahn, Martin Köbel, David Huntsman, Robert A. Soslow, Anna Defazio, Michael J. Birrer, Joe W. Gray, John N. Weinstein, David D. Bowtell, Ronny Drapkin, Jill P. Mesirov, Gad Getz, Douglas A. Levine, Matthew Meyerson




































Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)