Advertisement
Research ArticleGeneticsNeuroscience
Open Access | ![]() 10.1172/JCI191508
10.1172/JCI191508
Multimodal analysis of cell-free DNA identifies epigenetic biomarkers for amyotrophic lateral sclerosis diagnosis and progression
Sebastian Michels,1 Chaorong Chen,2 Wolfgang P. Ruf,1 M. Madhy Garcia Garcia,3 Frederick J. Arnold,3 Zhuoxing Wu,2 Craig L. Bennett,3 Daniel Shams,2 Leslie M. Thompson,2,4,5,6,7,8 Alyssa C. Walker,9 Dennis W. Dickson,9,10 Leonard Petrucelli,9,10 Johannes Dorst,1 Mercedes Prudencio,9,10 Wei Li,2 and Albert R. La Spada2,3,5,6,7,8
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Michels, S. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Chen, C. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Ruf, W. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Garcia Garcia, M. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Arnold, F. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Wu, Z. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Bennett, C. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Shams, D. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Thompson, L. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Walker, A. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by
Dickson, D.
in:
PubMed
|
Google Scholar
|

1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Petrucelli, L. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Dorst, J. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by
Prudencio, M.
in:
PubMed
|
Google Scholar
|
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by Li, W. in: PubMed | Google Scholar
1Department of Neurology, University of Ulm, Ulm, Germany.
2Department of Biological Chemistry,
3Department of Pathology & Laboratory Medicine,
4Department of Psychiatry & Human Behavior, and
5Department of Neurobiology & Behavior, University of California, Irvine, Irvine, California, USA.
6UCI Institute for Memory Impairment & Neurological Disorders, Irvine, California, USA.
7UCI Stem Cell Research Center, Irvine, California, USA.
8UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
9Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
10Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Irvine, California 92697, USA. Phone: 1.425.246.5779; Email: alaspada@uci.edu. Or to: Wei Li, Biological Chemistry, University of California Irvine, Irvine, California 92697, USA. Email: wei.li@uci.edu.
Authorship note: SM and CC contributed equally to this work.
Find articles by La Spada, A. in: PubMed | Google Scholar
Published June 1, 2026 - More info
J Clin Invest. 2026;136(11):e191508. https://doi.org/10.1172/JCI191508.
© 2026 Michels et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: January 22, 2025; Accepted: March 24, 2026

-
Results
Patients with ALS display a distinct methylome signature. We ascertained a study cohort consisting of 20 patients with sALS, 10 patients with C9-ALS, 10 presymptomatic carriers of C9-ALS, and people who are controls in the study and who are disease free (Table 1), with extensive clinical documentation of disease course and special studies, including measurement of neurofilaments. We isolated cfDNA from their banked serum samples, and after confirming that isolated cfDNA samples exhibit a peak distribution of DNA fragments migrating at approximately 170 bp (Figure 1A), we then performed targeted methylome sequencing on all 61 participants using the Enzymatic Methyl-seq (EM-seq) method, followed by unbiased target enrichment with the TWIST Human Methylome Panel of biotinylated probes that provide coverage of approximately 4 million CpG sites across the human genome (www.twistbioscience.com). With the exception of one sALS sample, we achieved satisfactory quality control and sufficient unique read depth at approximately 45× to permit in-depth epigenetic analysis of the resultant sequencing data for 60 samples, and we detected hundreds of de novo differentially methylated regions (DMRs) and associated genes between these 4 groups (Figure 1, B–E). To assess the importance of achieving approximately 45× sequencing depth, we conducted a simulation analysis and found that a substantial portion of DMRs would be missed at sequencing depths less than 45× (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI191508DS1).
 are detectable in plasma cell-free DNA obtained from patients with ALS and carriers.") Figure 1
Figure 1Numerous differentially methylated regions (DMRs) are detectable in plasma cell-free DNA obtained from patients with ALS and carriers. (A) Here, we see the size distribution of cell-free DNA obtained from 1.5 mL of blood serum. Note the major peak at approximately 170 bp that perfectly corresponds to the size of DNA protected by a single nucleosome and a minor peak for a dinucleosome protected DNA fragment at approximately 330 bp. (B–E) Volcano plots showing differentially methylated genes as a function of mean methylation difference (x axis) identified by DMR analysis of the following cohorts: (B) patients with sALS versus nondisease control individuals; (C) patients with C9-ALS versus patients with sALS; (D) patients with C9-ALS versus individuals without disease acting as controls; and (E) C9 patients with ALS versus carriers of C9-ALS. DMRs with an adjusted P value under 1 × 10–5 are colored.
When we extended our analysis to single CpG resolution, we identified 2,382 CpG sites with significantly different methylation between patients with sALS and people who were controls, and 1,801 CpG sites with significantly different methylation between carriers of C9-ALS and people who were controls. Clustering of the top 1,000 differentially methylated CpG sites (ranked by adjusted P values) for these cohort comparisons yielded clearly distinguishable methylation patterns (Figure 2, A and B).
 Figure 2
Figure 2Clustering of differentially methylated CpGs yields clear separation of groups of patients with ALS and individuals without disease acting as controls. (A and B) Clustering of patient groups based upon detection of differentially methylated CpGs in cfDNA samples subjected to targeted EM-seq. Here, we clustered the top 1,000 differentially methylated CpGs for: (A) patients with sALS versus nondisease control individuals, and (B) carriers of C9-ALS versus nondisease control individuals. Note that each column is a single sample and each row is a single CpG site. The threshold for significantly differentially methylated CpG sites was set at P < 0.001, based upon applying the Mann-Whitney U-test. (C) Tissue-of-origin results based upon UXM deconvolution analysis of cell-type–specific methylation markers that map to 7 different tissue types for each of the cohort groups. Note representation of different tissue types between patients with C9-ALS, carriers of the C9-HRE, patients with sALS, and nondisease control individuals is virtually identical, with approximately 4.5% of cfDNA derived from neural tissue for each cohort.
Deconvolution analysis reveals the origin of cfDNA. To determine the tissue of origin of cfDNA using tissue-specific methylation patterns (19), we applied the UXM algorithm to deconvolute our cfDNA samples into 39 different cell types (20), grouped into 7 broad tissue categories: blood immune cells, connective tissue, epithelium, sex-related tissue, neural tissue, muscle, and other. Our results revealed that approximately 4.5% of cfDNA is derived from neural tissue (Figure 2C and Supplemental Table 1), suggesting that a blood-based liquid biopsy can reliably detect methylation changes originating in the central nervous system (CNS). To better understand the origin of the neural cfDNA signal, we performed an expanded tissue deconvolution analysis incorporating brain cell subtypes (21) and determined that excitatory neurons are predominant contributors to neural-derived cfDNA (Supplemental Figure 2). Furthermore, our analysis of cfDNA tissue type did not reveal significant global differences in cfDNA tissue-of-origin among these cohorts. To assure accuracy of the deconvolution, we performed a simulation study using in silico mixtures of sequenced reads. For 50 mixtures at 5 different neuron sample concentrations varying from 0%–10%, the simulation results confirmed that the UXM algorithm can accurately detect cfDNA tissue-of-origin (Supplemental Figure 3).
Patients with ALS show cfDNA methylation alterations in disease-relevant genes. The most established biological mechanism for epigenetic gene regulation is the methylation status of the promoter region. While promoter hypermethylation typically leads to decreased gene expression, promoter hypomethylation results in increased gene expression. To further explore the relevance of differentially methylated genes in cfDNA, we applied a stringent filter to differentially methylated regions (DMRs) in the promoter region. Specifically, we retained promoter-overlapping DMRs with greater than or equal to 10 CpG sites, an adjusted P value less than 1 × 10–5 between groups, and a mean methylation difference greater than or equivalent to ± 0.1 between groups. DMRs in the X chromosome were excluded, as they naturally exhibit significant differences due to X-inactivation between the male and female sex. Unbiased de novo DMR analysis of patients with sALS versus nondisease control individuals revealed 18 genes with significant promoter methylation changes (Table 2). Of these highly significant hits, the DMR associated with PON1, the gene encoding Paraoxonase 1, exhibited the greatest change in methylation pattern. In the sALS cohort, the PON1 DMR was hypomethylated with an average promoter methylation of 43.6% compared with 57.6% in controls (P = 5.0 × 10–18). PON1 encodes an enzyme found in high-density lipoprotein (HDL) (22). While the association of ALS and cardiovascular disease is ambiguous, there is evidence that alterations in cholesterol metabolism are a risk factor and a negative prognostic factor in ALS (23, 24). Furthermore, several genetic single nucleotide polymorphism (SNP) coding variants in PON1 have been associated with increased susceptibility to sALS, including Q192R, L55M, and A162G (25–28), and another independent study reported a common haplotype in the PON1 promoter that is significantly linked to sALS disease risk (29).
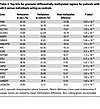 Table 2
Table 2Top hits for promoter differentially methylated regions for patients with sALS versus individuals acting as controls
Other ALS disease-relevant genes were also identified as displaying markedly different methylation patterns in promoter DMRs, including the LMTK3 gene that encodes the lemur tail kinase-3 protein and the STK11 gene that encodes the protein LKB1, a well-known tumor suppressor (Table 2). LMTK3 belongs to a family of lemur tyrosine kinases that are highly expressed in layer II to VI of the motor cortex and in the CA1-3 area in the dentate gyrus of the hippocampus (30). While LMTK3 normally functions in neurite extension and apoptosis, axonal transport, and endosomal vesicle trafficking (31, 32), LMTK3 can regulate cortical excitability through its interactions with the chloride-potassium transporter KCC2 (33), potentially implicating LMTK3 in excitotoxicity, which is a defining feature of ALS. Notably, postmortem studies of the frontal cortex of patients with sALS revealed increased expression of LMTK3 in neuron axons and dendrites (34), which could be explained by hypomethylation of the LMTK3 promoter DMR observed in our sALS patient cohort and found to be also present in C9-ALS presymptomatic carriers (Table 2). In the case of STK11, an unbiased integrative multi-omics study of sALS found that copy number variants (CNVs) in STK11 are associated with ALS disease risk and might represent a potential biomarker for sALS patient stratification (35), although the effect of the CNV increase at the STK11 locus upon gene expression was not determined. Whether hypermethylation of the STK11 promoter DMR detected by our methylome sequencing analysis of patients with sALS is related to the CNV increase observed at this locus in patients with sALS deserves consideration.
Since promoter methylation plays an important role in regulating gene expression, we further estimated the methylation level of promoter DMCs for ALS-associated genes (Figure 3). For TARDBP, which encodes the TDP-43 protein, we noted significant hypomethylation in both patients with sALS and patients with C9-ALS compared with nondisease control individuals, but remarkably, we observed no differences in hypomethylation between carriers of C9-ALS and nondisease control individuals (Figure 3), suggesting that epigenetic changes in the TDP-43 gene do not occur until after initiation of clinically significant disease. Interestingly, a postmortem analysis of the frontal cortex of a ALS patients found that TARDBP hypomethylation correlates with age of disease onset (36). Analysis of the C9orf72 gene revealed marked variability in methylation status, with markedly increased promoter methylation in patients with C9-ALS and carriers of C9-ALS (Figure 3), in agreement with prior studies (37). While there were trends towards CpG hypomethylation of SOD1 and AGRN in patients with C9-ALS and carriers, and evidence for CpG hypermethylation of FUS in patients with C9-ALS, these differences did not achieve statistical significance (Figure 3). We also noted marked promoter CpG hypomethylation of the ATXN2 gene in patients with C9-ALS, especially when compared with nondisease control individuals (Figure 3). These results indicate that ALS-linked genes are subject to CpG-level epigenetic alterations that can be detected in cfDNA isolated from serum of patients with sALS and C9-ALS.
 Figure 3
Figure 3CpG methylation levels in promoters of certain ALS-associated genes vary significantly between patients with sALS, patients with C9-ALS, carriers of C9-HRE, and individuals without disease acting as controls. Here we see a plot of the methylation levels for the top promoter DMCs of the listed ALS-associated genes measured in cfDNA samples obtained from blood plasma after normalization. The Y-axis corresponds with the methylation level, which is given as a Z score. For this comparison, a positive Z score means hypermethylation, while a negative Z score equates to hypomethylation. Statistical comparisons for significant differences are shown based upon applying the Mann-Whitney U test. *P < 0.05, **P < 0.01. Error bars indicate the SD.
Multimodal epigenetic sequencing analysis of cfDNA enables detection of patients with ALS. To permit a more comprehensive analysis of epigenetic signatures in cfDNA samples from patients with ALS, we extracted 3 additional epigenetic features from our methylome sequencing data: (a) DNase I hypersensitive site (DHS) region methylation, (b) nucleosome occupancy, and (c) window protection score. DHSs are established markers of regulatory DNA, which reveal novel relationships between chromatin accessibility, transcription, and DNA methylation (38); nucleosome occupancy reflects the likelihood that a given base pair in a cell will be occupied by a nucleosome; and window protection score is a measure of the dynamics of DNA fragmentation as it relates to nucleosome positioning (18). We have previously combined these 3 metrics with differential CpG site methylation to build a machine learning model for colorectal cancer detection, which we called multimodal epigenetic sequencing analysis (MESA) (39), to identify distinct epigenetic features for evaluation of methylome sequencing data from cfDNA. To determine if cfDNA-based epigenetic properties can distinguish patients with ALS from nondisease control individuals, we applied the MESA pipeline to our data set and trained a model on the most significant markers for these 4 modalities. To test the performance of MESA in detecting patients with sALS, we used leave-one-out cross-validation (LOOCV) and obtained an average area under the curve (AUC) of 0.91 in the receiver operator curve (ROC) characteristic analysis (Figure 4). Notably, with a false positive rate of 0% (i.e., 100% specificity), comparison of cfDNA epigenetic signatures enabled us to detect 70% of patients with sALS. To determine the robustness and generalizability of MESA, we considered a recently published cfDNA whole-genome bisulfite sequencing data set obtained from 10 patients with ALS and 8 nondisease control individuals (40). When we applied our MESA algorithm to this independent cohort, we obtained an AUC of 0.85, cross validating its strong performance for different sequencing methods and geographically diverse cohorts. We also inspected inter-individual biomarker variability for our top 100 differentially methylated CpG (DMC) sites, quantified by coefficient of variation, and noted that methylation variability was much greater in carriers of C9-ALS and patients with sALS compared with nondisease control individuals (Supplemental Table 2). This variability indicates that cfDNA methylation is capturing biological differences between different cohorts and shows that cfDNA-based biomarkers exhibit greater variability in disease and carrier groups.
 Figure 4
Figure 4MESA analysis can accurately detect patients with sALS in comparison with individuals without disease acting as controls. Using MESA, we performed receiver operator characteristic curve analysis for the most significantly different cfDNA multimodal epigenetic markers for patients with sALS (n = 19) versus individuals without disease acting as controls (n = 21). LOOCV analysis yielded an AUC of 0.91 and was then applied to an independent cross-validation cohort (Cross-cohort) of patients with sALS (n = 10) versus individuals without disease acting as controls (n = 8), yielding an AUC of 0.85.
CSF neurofilament levels and differential promoter methylation correlate with ALS disease progression. At present, there is an urgent need for reliable biomarkers to track disease progression in patients with ALS. The measurement of neurofilaments is being utilized as a biomarker in ALS, as neurofilaments may reflect axonal damage and ongoing motor neuron degeneration. We thus considered the utility of neurofilament levels as a predictor of disease progression in the 18 patients with sALS and 7 patients with C9-ALS from our study cohort or for whom neurofilament levels in blood and CSF have been obtained and longitudinal assessments had been performed. When we compared disease progression between these 18 patients with sALS and 7 patients with C9-ALS, we noted more rapid disease progression in patients with sALS, based upon a higher median loss of ALS-FRS-R points per month, as patients with sALS declined at an average rate of 0.80 ALS-FRS-R points per month, while patients with C9-ALS declined at an average rate of 0.67 ALS-FRS-R points per month, but this difference did not achieve statistical significance due to the small cohort sizes. In line with previous studies (41), the level of phosphorylated neurofilament heavy chain (pNfH) in CSF from our 18 patients with sALS and 7 patients with C9-ALS correlated with this difference in disease progression, with patients with sALS displaying higher median CSF pNfH levels in comparison with patients with C9-ALS (3,000 pg/mL vs. 2,202 pg/mL). However, these differences were not statistically significant, owing to the small cohort sizes. The sALS cohort and C9-ALS cohort were similar in terms of location of disease onset (approximately 80% spinal and approximately 20% bulbar; P = 0.949), age (P = 0.189), and sex (P = 0.135). When we evaluated CSF pNfH as a biomarker for ALS disease progression in all 25 patients with ALS, we observed a significant correlation between the slope of ALS-FRS-R decline and the increase in pNfH levels (Figure 5A). This data supports pNfH levels as an indicator of disease progression in our study cohort.
 Figure 5
Figure 5Neurofilament levels and disease progression rates correlate with single gene methylation changes in patients with ALS. (A) Plot of phosphorylated neurofilament heavy chain (pNfH) levels as a function of ALS-FRS-R rate of disease progression for 25 patients with ALS (sALS, n = 18; C9 ALS, n = 7). Statistical comparison is based upon Spearman rank correlation analysis. (B) Plot of phosphorylated neurofilament heavy chain (pNfH) levels as a function of RIPK1 gene methylation for patients with sALS (n = 18). Statistical comparison is based upon Spearman rank correlation analysis. (C) Plot of both phosphorylated neurofilament heavy chain (pNfH) levels and ALS-FRS-R rate of disease progression as a function of GBP1 gene methylation for patients with sALS (n = 18). Statistical comparison is based upon Spearman rank correlation analysis. (D) Plot of ALS-FRS-R rate of disease progression as a function of NDRG2 gene methylation for patients with sALS (n = 19). Statistical comparison is based upon Spearman rank correlation analysis. (E) Plot of ALS-FRS-R rate of disease progression as a function of FITM2 gene methylation for patients with sALS (n = 19). Statistical comparison is based upon Spearman rank correlation analysis.
We next tested if pNfH levels were correlated with promoter CpG site methylation differences in selected genes. Among genes with the most significant DMCs, we identified a negative correlation between pNfH CSF levels and methylation levels of the RIPK1 gene in patients with sALS (Figure 5B and Table 3). RIPK1 encodes the receptor-interacting serine/threonine protein kinase 1, is upregulated in postmortem spinal cord tissue from patients with ALS (42), and its serum concentrations were found to be higher in patients with ALS (43). In contrast, in the case of the GBP1 gene, we detected a positive correlation between pNfH CSF levels, and GBP1 methylation (Figure 5C and Table 3). GBP1 encodes a large GTPase involved in cellular signal transduction responses to inflammation and environmental stressors.
To further determine if CpG methylation differences in a single gene could be a significant predictor of ALS clinical disease progression, we next considered our top promoter differentially methylated CpG (DMC) hits. Interestingly, methylation of a GBP1 DMC also directly correlated with sALS clinical disease progression (Figure 5C and Table 4). We further noted that DMC methylation of the N-myc downstream-regulated gene 2 (NDRG2) exhibited a strong negative correlation with ALS-FRS-R slope (Figure 5D and Table 4), as patients with sALS defined as displaying faster disease progression have greatly reduced NDRG2 promoter CpG site methylation. The NDRG2 protein is known for its role as a tumor suppressor, inhibiting several key cell pathways, including Akt, NF-κB, and TGF-β signaling (44), but NDRG2 protein is also highly expressed in motor neurons (45), and can promote neurite outgrowth (46) and regulate oxidative stress. Interestingly, splicing dysregulation of the NDRG2 gene occurs upon knockdown of either FUS/TLS or TDP-43 (47), and increased NDRG2 protein expression, as would be expected upon promoter hypomethylation, has been implicated in disease pathogenesis in ALS-model mice (45). Finally, we noted that DMC methylation of FITM2, which encodes the fat storage–inducing transmembrane protein 2, displayed a strong negative correlation with clinical ALS disease progression (Figure 5E and Table 4). FIT proteins are critical for promoting the formation of lipid droplets within the endoplasmic reticulum. FITM2 is highly expressed in adipocytes, where it facilitates production of large lipid droplets essential for energy storage and metabolic homeostasis (48). Dysregulation of lipid droplets has been implicated in ALS disease pathogenesis, as TDP-43 loss of function results in metabolic disturbances, including an accumulation of lipid droplets (49). Furthermore, dramatically increased lipid droplet accumulation was detected in skeletal muscle biopsies obtained from patients with fALS carrying mutations in FUS (50).
Studies of ALS-associated RIPK1 hypomethylation confirm expression increase in human patients and implicate RIPK1 expression increase in microglia dysfunction. To determine if highly significant cfDNA promoter methylation differences found to correlate with ALS patient CSF neurofilament burden or disease severity result in altered expression of the implicated genes, we selected NDRG2 and RIPK1 for further analysis, as these 2 target genes are known to be dysregulated in ALS model mice and in human patients (42, 43, 45). When we measured the RNA expression levels of NDRG2 and RIPK1 in the frontal cortex of patients with frontotemporal dementia associated with TDP-43 (FTD) with or without ALS motor neuron disease, we noted increased expression of both these genes in FTD-TDP and patients with FTD-TDP/ALS, though the increase only constituted a trend for NDRG2 (Figure 6A). A previous single-nucleus RNA sequencing study on sALS spinal cord showed that increased expression of RIPK1 in glial cells is associated with neuroinflammation (51). To validate whether increased RIPK1 expression leads to inflammation in microglia, we transduced induced microglia-like cells (iTF-microglia), derived from induced pluripotent stem cells (52), with a RIPK1 lentivirus and stimulated inflammation by treating with lipopolysaccharide (LPS). We observed a robust cytokine response, which was significantly augmented by increased expression of RIPK1 (Figure 6B). We then assayed the phagocytosis capacity of iTF-microglia subjected to LPS treatment and RIPK1 expression increase, and we found that increased RIPK1 significantly impaired phagocytosis (Figure 6C), indicating that RIPK1 diminished this neuroprotective function. Another key microglia task is migration, which is involved in the response to injury. To assay microglia migration, which is accentuated when microglia are reactive, we treated iTF-microglia with LPS in the presence or absence of RIPK1, and we observed markedly increased iTF-microglia migration with RIPK1 overexpression (Figure 6D). Furthermore, higher RIPK1 expression led to enhanced cell death (Figure 6E). Overall, these findings support a role for increased RIPK1 expression in promoting glial cell dysfunction and neuroinflammation.
 Figure 6
Figure 6Expression levels of differentially methylated genes in postmortem FTD-ALS patient cortex and evaluation of effect of RIPK1 expression increase on microglia function. (A) Quantitative real-time RT-PCR analysis of RNAs isolated from the frontal cortex of patients with FTD (n = 167) or patients with FTD-ALS (n = 59) with evidence of TDP-43 histopathology on postmortem examination, as well as individuals without disease acting as controls (n = 51) to measure the expression level of NDGR2 and RIPK1. Statistical significance was calculated using the Kruskal-Wallis nonparametric test. *P < 0.05, ****P < 0.0001. (B) iTF-microglia were transduced with a RIPK1 lentivirus expression or empty vector, then left untreated or subjected to LPS treatment. We then isolated RNAs and performed qRT-PCR analysis of the indicated cytokine genes. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.001; ANOVA with Tukey’s post hoc test, n = 3 biological replicates. (C) iTF-microglia were transduced with a RIPK1 lentivirus expression or empty vector, and then left untreated or subjected to LPS treatment. We then incubated iTF-microglia with pHrodo Red Zymosan BioParticles Conjugate, and measured pHrodo Red Zymosan BioParticles fluorescence as a ratio of DAPI fluorescence. *P < 0.05, **P < 0.01; ANOVA with Tukey’s post hoc test, n = 4 biological replicates. (D) iTF-microglia were transduced with a RIPK1 lentivirus expression or empty vector, then left untreated or subjected to LPS treatment. To measure migration, iTF-Microglia were plated onto PDL and ECM coated 8 μm transwells, ADP was added as the attractant, and numbers of migrating microglia were counted, *P < 0.05, ****P < 0.001; ANOVA with Tukey’s post hoc test, n = 6 biological replicates. Boxes indicate the 25th percentile to 75th percentile with respective medians marked by a line within the box, while whiskers encompass the full range of scores for each condition. Please see the supporting raw values file for exact scores for each condition. (E) iTF-microglia were transduced with a RIPK1 lentivirus expression or empty vector, and then left untreated or subjected to LPS treatment. We then stained iTF-microglia with DAPI and counted the numbers of dead cells based on amoeboid and blebbing appearance. *P < 0.05, ***P < 0.001, ****P < 0.0001; ANOVA with Tukey’s post hoc test, n = 4 biological replicates. Error bars indicate SEM.
 are detectable in plasma cell-free DNA obtained from patients with ALS and carriers.")












Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)