Review

Abstract
Recent data underscore the importance of intertissue communication in the maintenance of normal glucose homeostasis. Important signals are conveyed by hormones, cytokines, and fuel substrates and are sensed through a variety of cellular mechanisms. The ability of tissues to sense and adapt to changes in metabolic status and fuel availability is altered in insulin-resistant states including type 2 diabetes. Here we review the roles of glucose and its metabolites as signaling molecules and the diverse physiologic mechanisms for glucose sensing.
Authors
Mark A. Herman, Barbara B. Kahn

Abstract
AMP-activated protein kinase (AMPK) is an energy sensor that regulates cellular metabolism. When activated by a deficit in nutrient status, AMPK stimulates glucose uptake and lipid oxidation to produce energy, while turning off energy-consuming processes including glucose and lipid production to restore energy balance. AMPK controls whole-body glucose homeostasis by regulating metabolism in multiple peripheral tissues, such as skeletal muscle, liver, adipose tissues, and pancreatic β cells — key tissues in the pathogenesis of type 2 diabetes. By responding to diverse hormonal signals including leptin and adiponectin, AMPK serves as an intertissue signal integrator among peripheral tissues, as well as the hypothalamus, in the control of whole-body energy balance.
Authors
Yun Chau Long, Juleen R. Zierath

Abstract
Insulin has pleiotropic biological effects in virtually all tissues. However, the relevance of insulin signaling in peripheral tissues has been studied far more extensively than its role in the brain. An evolving body of evidence indicates that in the brain, insulin is involved in multiple regulatory mechanisms including neuronal survival, learning, and memory, as well as in regulation of energy homeostasis and reproductive endocrinology. Here we review insulin’s role as a central homeostatic signal with regard to energy and glucose homeostasis and discuss the mechanisms by which insulin communicates information about the body’s energy status to the brain. Particular emphasis is placed on the controversial current debate about the similarities and differences between hypothalamic insulin and leptin signaling at the molecular level.
Authors
Leona Plum, Bengt F. Belgardt, Jens C. Brüning
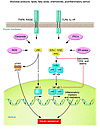
Abstract
Over a hundred years ago, high doses of salicylates were shown to lower glucose levels in diabetic patients. This should have been an important clue to link inflammation to the pathogenesis of type 2 diabetes (T2D), but the antihyperglycemic and antiinflammatory effects of salicylates were not connected to the pathogenesis of insulin resistance until recently. Together with the discovery of an important role for tissue macrophages, these new findings are helping to reshape thinking about how obesity increases the risk for developing T2D and the metabolic syndrome. The evolving concept of insulin resistance and T2D as having immunological components and an improving picture of how inflammation modulates metabolism provide new opportunities for using antiinflammatory strategies to correct the metabolic consequences of excess adiposity.
Authors
Steven E. Shoelson, Jongsoon Lee, Allison B. Goldfine

Abstract
The major focus of this Review is on the mechanisms of islet β cell failure in the pathogenesis of obesity-associated type 2 diabetes (T2D). As this demise occurs within the context of β cell compensation for insulin resistance, consideration is also given to the mechanisms involved in the compensation process, including mechanisms for expansion of β cell mass and for enhanced β cell performance. The importance of genetic, intrauterine, and environmental factors in the determination of “susceptible” islets and overall risk for T2D is reviewed. The likely mechanisms of β cell failure are discussed within the two broad categories: those with initiation and those with progression roles.
Authors
Marc Prentki, Christopher J. Nolan

Abstract
Adiponectin is an adipokine that is specifically and abundantly expressed in adipose tissue and directly sensitizes the body to insulin. Hypoadiponectinemia, caused by interactions of genetic factors such as SNPs in the Adiponectin gene and environmental factors causing obesity, appears to play an important causal role in insulin resistance, type 2 diabetes, and the metabolic syndrome, which are linked to obesity. The adiponectin receptors, AdipoR1 and AdipoR2, which mediate the antidiabetic metabolic actions of adiponectin, have been cloned and are downregulated in obesity-linked insulin resistance. Upregulation of adiponectin is a partial cause of the insulin-sensitizing and antidiabetic actions of thiazolidinediones. Therefore, adiponectin and adiponectin receptors represent potential versatile therapeutic targets to combat obesity-linked diseases characterized by insulin resistance. This Review describes the pathophysiology of adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome.
Authors
Takashi Kadowaki, Toshimasa Yamauchi, Naoto Kubota, Kazuo Hara, Kohjiro Ueki, Kazuyuki Tobe

Abstract
It is now well accepted that diabetes mellitus is one of the main threats to human health in the twenty-first century. The total number of people with diabetes worldwide was estimated at between 151 million and 171 million in 2000 and is projected to increase to 221 million in 2010 and to 366 million in 2030. Needless to say, the increase in the number of people with diabetes will be accompanied by an increase in the number of those with diabetic complications such as nephropathy, retinopathy, neuropathy, and atherosclerosis. The global mortality attributable to diabetes in the year 2000 was estimated at 2.9 million deaths, a number that will also increase. Given that type 2 diabetes accounts for more than 90% of cases of diabetes worldwide, it is important that we understand the pathogenesis of this condition and develop new approaches to its prevention and treatment.
Authors
Masato Kasuga

Abstract
Skin is at the interface between the complex physiology of the body and the external, often hostile, environment, and the semipermeable epidermal barrier prevents both the escape of moisture and the entry of infectious or toxic substances. Newborns with rare congenital barrier defects underscore the skin’s essential role in a terrestrial environment and demonstrate the compensatory responses evoked ex utero to reestablish a barrier. Common inflammatory skin disorders such as atopic dermatitis and psoriasis exhibit decreased barrier function, and recent studies suggest that the complex response of epidermal cells to barrier disruption may aggravate, maintain, or even initiate such conditions. Either aiding barrier reestablishment or dampening the epidermal stress response may improve the treatment of these disorders. This Review discusses the molecular regulation of the epidermal barrier as well as causes and potential treatments for defects of barrier formation and proposes that medical management of barrier disruption may positively affect the course of common skin disorders.
Authors
Julia A. Segre

Abstract
Autoimmune bullous disorders are a group of severe skin diseases characterized clinically by blisters and erosions of skin and/or mucous membranes. A hallmark of these disorders is the presence of IgG and occasionally IgA autoantibodies that target distinct adhesion structures of the epidermis, dermoepidermal basement membrane, and anchoring fibrils of the dermis. This Review focuses on the potential role of autoreactive T cells in the pathogenesis of these disorders. Pemphigus vulgaris (PV) and bullous pemphigoid (BP) are the best-characterized bullous disorders with regard to pathogenesis and T cell involvement. Activation of autoreactive T cells in PV and BP is restricted by distinct HLA class II alleles that are prevalent in individuals with these disorders. Autoreactive T cells are not only present in patients but can also be detected in healthy individuals. Recently, a subset of autoreactive T cells with remarkable regulatory function was identified in healthy individuals and to a much lesser extent in patients with PV, suggesting that the occurrence of autoimmune bullous disorders may be linked to a dysfunction of Tregs.
Authors
Michael Hertl, Rüdiger Eming, Christian Veldman

Abstract
Wnt proteins are a family of secreted proteins that regulate many aspects of cell growth, differentiation, function, and death. Considerable progress has been made in our understanding of the molecular links between Wnt signaling and bone development and remodeling since initial reports that mutations in the Wnt coreceptor low-density lipoprotein receptor–related protein 5 (LRP5) are causally linked to alterations in human bone mass. Of the pathways activated by Wnts, it is signaling through the canonical (i.e., Wnt/β-catenin) pathway that increases bone mass through a number of mechanisms including renewal of stem cells, stimulation of preosteoblast replication, induction of osteoblastogenesis, and inhibition of osteoblast and osteocyte apoptosis. This pathway is an enticing target for developing drugs to battle skeletal diseases as Wnt/β-catenin signaling is composed of a series of molecular interactions that offer potential places for pharmacological intervention. In considering opportunities for anabolic drug discovery in this area, one must consider multiple factors, including (a) the roles of Wnt signaling for development, remodeling, and pathology of bone; (b) how pharmacological interventions that target this pathway may specifically treat osteoporosis and other aspects of skeletal health; and (c) whether the targets within this pathway are amenable to drug intervention. In this Review we discuss the current understanding of this pathway in terms of bone biology and assess whether targeting this pathway might yield novel therapeutics to treat typical bone disorders.
Authors
Venkatesh Krishnan, Henry U. Bryant, Ormond A. MacDougald
No posts were found with this tag.



Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)