Concise Communications

Abstract
Targeted cancer therapies, which act on specific cancer-associated molecular targets, are predominantly inhibitors of oncogenic kinases. While these drugs have achieved some clinical success, the inactivation of kinase signaling via stimulation of endogenous phosphatases has received minimal attention as an alternative targeted approach. Here, we have demonstrated that activation of the tumor suppressor protein phosphatase 2A (PP2A), a negative regulator of multiple oncogenic signaling proteins, is a promising therapeutic approach for the treatment of cancers. Our group previously developed a series of orally bioavailable small molecule activators of PP2A, termed SMAPs. We now report that SMAP treatment inhibited the growth of KRAS-mutant lung cancers in mouse xenografts and transgenic models. Mechanistically, we found that SMAPs act by binding to the PP2A Aα scaffold subunit to drive conformational changes in PP2A. These results show that PP2A can be activated in cancer cells to inhibit proliferation. Our strategy of reactivating endogenous PP2A may be applicable to the treatment of other diseases and represents an advancement toward the development of small molecule activators of tumor suppressor proteins.
Authors
Jaya Sangodkar, Abbey Perl, Rita Tohme, Janna Kiselar, David B. Kastrinsky, Nilesh Zaware, Sudeh Izadmehr, Sahar Mazhar, Danica D. Wiredja, Caitlin M. O’Connor, Divya Hoon, Neil S. Dhawan, Daniela Schlatzer, Shen Yao, Daniel Leonard, Alain C. Borczuk, Giridharan Gokulrangan, Lifu Wang, Elena Svenson, Caroline C. Farrington, Eric Yuan, Rita A. Avelar, Agnes Stachnik, Blake Smith, Vickram Gidwani, Heather M. Giannini, Daniel McQuaid, Kimberly McClinch, Zhizhi Wang, Alice C. Levine, Rosalie C. Sears, Edward Y. Chen, Qiaonan Duan, Manish Datt, Shozeb Haider, Avi Ma’ayan, Analisa DiFeo, Neelesh Sharma, Matthew D. Galsky, David L. Brautigan, Yiannis A. Ioannou, Wenqing Xu, Mark R. Chance, Michael Ohlmeyer, Goutham Narla

Abstract
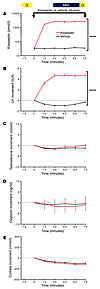
Accumulation of amyloid-β (Aβ) protein may cause synapse degeneration and cognitive impairment in Alzheimer’s disease (AD) by reactivating expression of the developmental synapse repressor protein Ephexin5 (also known as ARHGEF15). Here, we have reported that Aβ is sufficient to acutely promote the production of Ephexin5 in mature hippocampal neurons and in mice expressing human amyloid precursor protein (hAPP mice), a model for familial AD that produces high brain levels of Aβ. Ephexin5 expression was highly elevated in the hippocampi of human AD patients, indicating its potential relevance to AD. We also observed elevated Ephexin5 expression in the hippocampi of hAPP mice. Removal of Ephexin5 expression eliminated hippocampal dendritic spine loss and rescued AD-associated behavioral deficits in the hAPP mice. Furthermore, selective reduction of Ephexin5 expression using shRNA in the dentate gyrus of presymptomatic adolescent hAPP mice was sufficient to protect these mice from developing cognitive impairment. Thus, pathological elevation of Ephexin5 expression critically drives Aβ-induced memory impairment, and strategies aimed at reducing Ephexin5 levels may represent an effective approach to treating AD.
Authors
Gabrielle L. Sell, Thomas B. Schaffer, Seth S. Margolis
Abstract
Authors
Alexander N. Comninos, Matthew B. Wall, Lysia Demetriou, Amar J. Shah, Sophie A. Clarke, Shakunthala Narayanaswamy, Alexander Nesbitt, Chioma Izzi-Engbeaya, Julia K. Prague, Ali Abbara, Risheka Ratnasabapathy, Victoria Salem, Gurjinder M. Nijher, Channa N. Jayasena, Mark Tanner, Paul Bassett, Amrish Mehta, Eugenii A. Rabiner, Christoph Hönigsperger, Meire Ribeiro Silva, Ole Kristian Brandtzaeg, Elsa Lundanes, Steven Ray Wilson, Rachel C. Brown, Sarah A. Thomas, Stephen R. Bloom, Waljit S. Dhillo

Abstract
The mechanism by which leptin reverses diabetic ketoacidosis (DKA) is unknown. We examined the acute insulin-independent effects of leptin replacement therapy in a streptozotocin-induced rat model of DKA. Leptin infusion reduced rates of lipolysis, hepatic glucose production (HGP), and hepatic ketogenesis by 50% within 6 hours and were independent of any changes in plasma glucagon concentrations; these effects were abrogated by coinfusion of corticosterone. Treating leptin- and corticosterone-infused rats with an adipose triglyceride lipase inhibitor blocked corticosterone-induced increases in plasma glucose concentrations and rates of HGP and ketogenesis. Similarly, adrenalectomized type 1 diabetic (T1D) rats exhibited decreased rates of lipolysis, HGP, and ketogenesis; these effects were reversed by corticosterone infusion. Leptin-induced decreases in lipolysis, HGP, and ketogenesis in DKA were also nullified by relatively small increases (15 to 70 pM) in plasma insulin concentrations. In contrast, the chronic glucose-lowering effect of leptin in a STZ-induced mouse model of poorly controlled T1D was associated with decreased food intake, reduced plasma glucagon and corticosterone concentrations, and decreased ectopic lipid (triacylglycerol/diacylglycerol) content in liver and muscle. Collectively, these studies demonstrate marked differences in the acute insulin-independent effects by which leptin reverses fasting hyperglycemia and ketoacidosis in a rodent model of DKA versus the chronic pleotropic effects by which leptin reverses hyperglycemia in a non-DKA rodent model of T1D.
Authors
Rachel J. Perry, Liang Peng, Abudukadier Abulizi, Lynn Kennedy, Gary W. Cline, Gerald I. Shulman

Abstract
Despite the efficient suppression of HIV-1 replication that can be achieved with combined antiretroviral therapy (cART), low levels of type I interferon (IFN-I) signaling persist in some individuals. This sustained signaling may impede immune recovery and foster viral persistence. Here we report studies using a monoclonal antibody to block IFN-α/β receptor (IFNAR) signaling in humanized mice (hu-mice) that were persistently infected with HIV-1. We discovered that effective cART restored the number of human immune cells in HIV-1–infected hu-mice but did not rescue their immune hyperactivation and dysfunction. IFNAR blockade fully reversed HIV-1–induced immune hyperactivation and rescued anti–HIV-1 immune responses in T cells from HIV-1–infected hu-mice. Finally, we found that IFNAR blockade in the presence of cART reduced the size of HIV-1 reservoirs in lymphoid tissues and delayed HIV-1 rebound after cART cessation in the HIV-1–infected hu-mice. We conclude that low levels of IFN-I signaling contribute to HIV-1–associated immune dysfunction and foster HIV-1 persistence in cART-treated hosts. Our results suggest that blocking IFNAR may provide a potential strategy to enhance immune recovery and reduce HIV-1 reservoirs in individuals with sustained elevations in IFN-I signaling during suppressive cART.
Authors
Liang Cheng, Jianping Ma, Jingyun Li, Dan Li, Guangming Li, Feng Li, Qing Zhang, Haisheng Yu, Fumihiko Yasui, Chaobaihui Ye, Li-Chung Tsao, Zhiyuan Hu, Lishan Su, Liguo Zhang

Abstract
Huntington’s disease (HD) is a polyglutamine disorder caused by a CAG expansion in the Huntingtin (
Authors
Laura Rué, Mónica Bañez-Coronel, Jordi Creus-Muncunill, Albert Giralt, Rafael Alcalá-Vida, Gartze Mentxaka, Birgit Kagerbauer, M. Teresa Zomeño-Abellán, Zeus Aranda, Veronica Venturi, Esther Pérez-Navarro, Xavier Estivill, Eulàlia Martí

Abstract
Enhancing energy expenditure (EE) is an attractive strategy to combat obesity and diabetes. Global deletion of
Authors
Qingzhang Zhu, Sarbani Ghoshal, Ana Rodrigues, Su Gao, Alice Asterian, Theodore M. Kamenecka, James C. Barrow, Anutosh Chakraborty

Abstract
Authors
Adil I. Daud, Kimberly Loo, Mariela L. Pauli, Robert Sanchez-Rodriguez, Priscila Munoz Sandoval, Keyon Taravati, Katy Tsai, Adi Nosrati, Lorenzo Nardo, Michael D. Alvarado, Alain P. Algazi, Miguel H. Pampaloni, Iryna V. Lobach, Jimmy Hwang, Robert H. Pierce, Iris K. Gratz, Matthew F. Krummel, Michael D. Rosenblum

Abstract
Autonomous thyroid adenomas (ATAs) are a frequent cause of hyperthyroidism. Mutations in the genes encoding the TSH receptor (
Authors
Davide Calebiro, Elisa S. Grassi, Markus Eszlinger, Cristina L. Ronchi, Amod Godbole, Kerstin Bathon, Fabiana Guizzardi, Tiziana de Filippis, Knut Krohn, Holger Jaeschke, Thomas Schwarzmayr, Rifat Bircan, Hulya Iliksu Gozu, Seda Sancak, Marek Niedziela, Tim M. Strom, Martin Fassnacht, Luca Persani, Ralf Paschke

Abstract
The structural maintenance of chromosomes (SMC) family of proteins supports mitotic proliferation, meiosis, and DNA repair to control genomic stability. Impairments in chromosome maintenance are linked to rare chromosome breakage disorders. Here, we have identified a chromosome breakage syndrome associated with severe lung disease in early childhood. Four children from two unrelated kindreds died of severe pulmonary disease during infancy following viral pneumonia with evidence of combined T and B cell immunodeficiency. Whole exome sequencing revealed biallelic missense mutations in the
Authors
Saskia N. van der Crabben, Marije P. Hennus, Grant A. McGregor, Deborah I. Ritter, Sandesh C.S. Nagamani, Owen S. Wells, Magdalena Harakalova, Ivan K. Chinn, Aaron Alt, Lucie Vondrova, Ron Hochstenbach, Joris M. van Montfrans, Suzanne W. Terheggen-Lagro, Stef van Lieshout, Markus J. van Roosmalen, Ivo Renkens, Karen Duran, Isaac J. Nijman, Wigard P. Kloosterman, Eric Hennekam, Jordan S. Orange, Peter M. van Hasselt, David A. Wheeler, Jan J. Palecek, Alan R. Lehmann, Antony W. Oliver, Laurence H. Pearl, Sharon E. Plon, Johanne M. Murray, Gijs van Haaften
No posts were found with this tag.



Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)