Review Series

Abstract
Human reproduction is relatively inefficient. Nearly 30% of pregnancies result in spontaneous losses, which are both a clinical problem and a psychological stress to the families involved. Furthermore, although the human population is growing rapidly and is predicted to reach 9 billion by 2050, 15% of couples worldwide are childless because of infertility. Many underlying causes of infertility have been overcome by assisted reproductive technologies such as in vitro fertilization, yet pregnancy success rates using such approaches remain disappointingly low. Since mechanistic approaches to study human reproductive processes are ethically restricted, future advances in fertility treatment and the development of new contraceptives rely predominantly on the study of the factors influencing reproduction in model systems. The articles in this Reproductive Biology Review series present updates on the current understanding of various reproductive processes in model systems and raise questions that need to be addressed if we are to improve human reproductive health.
Authors
Sudhansu K. Dey

Abstract
Mammalian preimplantation development, which is the period extending from fertilization to implantation, results in the formation of a blastocyst with three distinct cell lineages. Only one of these lineages, the epiblast, contributes to the embryo itself, while the other two lineages, the trophectoderm and the primitive endoderm, become extra-embryonic tissues. Significant gains have been made in our understanding of the major events of mouse preimplantation development, and recent discoveries have shed new light on the establishment of the three blastocyst lineages. What is less clear, however, is how closely human preimplantation development mimics that in the mouse. A greater understanding of the similarities and differences between mouse and human preimplantation development has implications for improving assisted reproductive technologies and for deriving human embryonic stem cells.
Authors
Katie Cockburn, Janet Rossant

Abstract
Much of our knowledge of human uterine physiology and pathology has been extrapolated from the study of diverse animal models, as there is no ideal system for studying human uterine biology in vitro. Although it remains debatable whether mouse models are the most suitable system for investigating human uterine function(s), gene-manipulated mice are considered by many the most useful tool for mechanistic analysis, and numerous studies have identified many similarities in female reproduction between the two species. This Review brings together information from studies using animal models, in particular mouse models, that shed light on normal and pathologic aspects of uterine biology and pregnancy complications.
Authors
Hyunjung Jade Lim, Haibin Wang

Abstract
The placenta provides critical transport functions between the maternal and fetal circulations during intrauterine development. Formation of this interface relies on coordinated interactions among transcriptional, epigenetic, and environmental factors. Here we describe these mechanisms in the context of the differentiation of placental cells (trophoblasts) and synthesize current knowledge about how they interact to generate a functional placenta. Developing an understanding of these pathways contributes to an improvement of our models for studying trophoblast biology and sheds light on the etiology of pregnancy complications and the in utero programming of adult diseases.
Authors
Emin Maltepe, Anna I. Bakardjiev, Susan J. Fisher
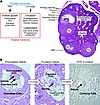
Abstract
The classical view of ovarian follicle development is that it is regulated by the hypothalamic-pituitary-ovarian axis, in which gonadotropin-releasing hormone (GnRH) controls the release of the gonadotropic hormones follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and that ovarian steroids exert both negative and positive regulatory effects on GnRH secretion. More recent studies in mice and humans indicate that many other intra-ovarian signaling cascades affect follicular development and gonadotropin action in a stage- and context-specific manner. As we discuss here, mutant mouse models and clinical evidence indicate that some of the most powerful intra-ovarian regulators of follicular development include the TGF-β/SMAD, WNT/FZD/β-catenin, and RAS/ERK1/2 signaling pathways and the FOXO/FOXL2 transcription factors.
Authors
JoAnne S. Richards, Stephanie A. Pangas

Abstract
Mammalian fertilization comprises sperm migration through the female reproductive tract, biochemical and morphological changes to sperm, and sperm-egg interaction in the oviduct. Recent gene knockout approaches in mice have revealed that many factors previously considered important for fertilization are largely dispensable, or if they are essential, they have an unexpected function. These results indicate that what has been observed in in vitro fertilization (IVF) differs significantly from what occurs during “physiological” fertilization. This Review focuses on the advantages of studying fertilization using gene-manipulated animals and highlights an emerging molecular mechanism of mammalian fertilization.
Authors
Masahito Ikawa, Naokazu Inoue, Adam M. Benham, Masaru Okabe

Abstract
Oocytes play a pivotal role in the cycle of human life. As we discuss here, after emerging from germline stem cells in the fetus, they grow in a follicular niche in which development is harmonized for timely ovulation and hormone secretion after puberty. Most human oocytes have poor developmental competence and are peculiarly vulnerable to chromosomal malsegregation, especially as women pass the optimal years of fertility and may begin to turn to assisted reproductive technologies (ARTs) and egg donation. Research needs to focus on the molecular factors involved and the environmental niche required for optimal development of oocytes, with the aim of increasing their numbers and quality for ARTs, since these are the factors that so often limit human fertility.
Authors
Roger Gosden, Bora Lee

Abstract
The ability to create new functional cardiomyocytes is the holy grail of cardiac regenerative medicine. From studies using model organisms, new insights into the fundamental pathways that drive heart muscle regeneration have begun to arise as well as a growing knowledge of the distinct families of multipotent cardiovascular progenitors that generate diverse lineages during heart development. In this Review, we highlight this intersection of the “pregenerative” biology of heart progenitor cells and heart regeneration and discuss the longer term challenges and opportunities in moving toward a therapeutic goal of regenerative cardiovascular medicine.
Authors
B. Alexander Yi, Oliver Wernet, Kenneth R. Chien

Abstract
Induced pluripotent stem (iPS) cells are generated by epigenetic reprogramming of somatic cells through the exogenous expression of transcription factors. These cells, just like embryonic stem cells, are likely to have a major impact on regenerative medicine, because they self-renew and retain the potential to be differentiated into all cell types of the human body. In this Review, we describe the current state of iPS cell technology, including approaches by which they are generated and what is known about their biology, and discuss the potential applications of these cells for disease modeling, drug discovery, and, eventually, cell replacement therapy.
Authors
Evangelos Kiskinis, Kevin Eggan

Abstract
Cancer stem cells (CSCs) are a subpopulation of tumor cells that selectively possess tumor initiation and self-renewal capacity and the ability to give rise to bulk populations of nontumorigenic cancer cell progeny through differentiation. As we discuss here, they have been prospectively identified in several human malignancies, and their relative abundance in clinical cancer specimens has been correlated with malignant disease progression in human patients. Furthermore, recent findings suggest that clinical cancer progression driven by CSCs may contribute to the failure of existing therapies to consistently eradicate malignant tumors. Therefore, CSC-directed therapeutic approaches might represent translationally relevant strategies to improve clinical cancer therapy, in particular for those malignancies that are currently refractory to conventional anticancer agents directed predominantly at tumor bulk populations.
Authors
Natasha Y. Frank, Tobias Schatton, Markus H. Frank
No posts were found with this tag.



Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)