Research Article

Abstract
The lymphatic vasculature is essential for maintaining interstitial fluid homeostasis, and dysfunctional lymphangiogenesis contributes to various pathological processes, including inflammatory disease and tumor metastasis. Mutations in
Authors
Anees Fatima, Ying Wang, Yutaka Uchida, Pieter Norden, Ting Liu, Austin Culver, William H. Dietz, Ford Culver, Meredith Millay, Yoh-suke Mukouyama, Tsutomu Kume

Abstract
Successful bacterial pathogens produce an array of virulence factors that allow subversion of the immune system and persistence within the host. For example, uropathogenic
Authors
Anna Waldhuber, Manoj Puthia, Andreas Wieser, Christine Cirl, Susanne Dürr, Silke Neumann-Pfeifer, Simone Albrecht, Franziska Römmler, Tina Müller, Yunji Zheng, Sören Schubert, Olaf Groß, Catharina Svanborg, Thomas Miethke

Abstract
Loss of the growth-suppressive effects of bone morphogenetic protein (BMP) signaling has been demonstrated to promote pulmonary arterial endothelial cell dysfunction and induce pulmonary arterial smooth muscle cell (PASMC) proliferation, leading to the development of pulmonary arterial hypertension (PAH). MicroRNAs (miRs) mediate higher order regulation of cellular function through coordinated modulation of mRNA targets; however, miR expression is altered by disease development and drug therapy. Here, we examined treatment-naive patients and experimental models of PAH and identified a reduction in the levels of miR-140-5p. Inhibition of miR-140-5p promoted PASMC proliferation and migration in vitro. In rat models of PAH, nebulized delivery of miR-140-5p mimic prevented the development of PAH and attenuated the progression of established PAH. Network and pathway analysis identified SMAD-specific E3 ubiquitin protein ligase 1 (SMURF1) as a key miR-140-5p target and regulator of BMP signaling. Evaluation of human tissue revealed that SMURF1 is increased in patients with PAH. miR-140-5p mimic or SMURF1 knockdown in PASMCs altered BMP signaling, further supporting these factors as regulators of BMP signaling. Finally,
Authors
Alexander M.K. Rothman, Nadine D. Arnold, Josephine A. Pickworth, James Iremonger, Loredana Ciuclan, Robert Allen, Sabine Guth-Gundel, Mark Southwood, Nicholas W. Morrell, Matthew Thomas, Sheila E. Francis, David J. Rowlands, Allan Lawrie
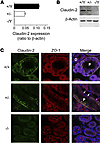
Abstract
Efficient oxygen utilization in the kidney may be supported by paracellular epithelial transport, a form of passive diffusion that is driven by preexisting transepithelial electrochemical gradients. Claudins are tight-junction transmembrane proteins that act as paracellular ion channels in epithelial cells. In the proximal tubule (PT) of the kidney, claudin-2 mediates paracellular sodium reabsorption. Here, we used murine models to investigate the role of claudin-2 in maintaining energy efficiency in the kidney. We found that claudin-2–null mice conserve sodium to the same extent as WT mice, even during profound dietary sodium depletion, as a result of the upregulation of transcellular Na-K-2Cl transport activity in the thick ascending limb of Henle. We hypothesized that shifting sodium transport to transcellular pathways would lead to increased whole-kidney oxygen consumption. Indeed, compared with control animals, oxygen consumption in the kidneys of claudin-2–null mice was markedly increased, resulting in medullary hypoxia. Furthermore, tubular injury in kidneys subjected to bilateral renal ischemia-reperfusion injury was more severe in the absence of claudin-2. Our results indicate that paracellular transport in the PT is required for efficient utilization of oxygen in the service of sodium transport. We speculate that paracellular permeability may have evolved as a general strategy in epithelial tissues to maximize energy efficiency.
Authors
Lei Pei, Glenn Solis, Mien T.X. Nguyen, Nikhil Kamat, Lynn Magenheimer, Min Zhuo, Jiahua Li, Joshua Curry, Alicia A. McDonough, Timothy A. Fields, William J. Welch, Alan S.L. Yu

Abstract
The molecular mechanisms that underlie spleen development and congenital asplenia, a condition linked to increased risk of overwhelming infections, remain largely unknown. The transcription factor TLX1 controls cell fate specification and organ expansion during spleen development, and
Authors
Elisa Lenti, Diego Farinello, Kazunari K. Yokoyama, Dmitry Penkov, Laura Castagnaro, Giovanni Lavorgna, Kenly Wuputra, Lisa L. Sandell, Naomi E. Butler Tjaden, Francesca Bernassola, Nicoletta Caridi, Anna De Antoni, Michael Wagner, Katja Kozinc, Karen Niederreither, Francesco Blasi, Diego Pasini, Gregor Majdic, Giovanni Tonon, Paul A. Trainor, Andrea Brendolan

Abstract
The histone demethylase PHF8 has been implicated in multiple pathological disorders, including X-linked mental retardation and tumorigenesis. However, it is not clear how the abundance and function of PHF8 are regulated. Here, we report that PHF8 physically associates with the deubiquitinase USP7. Specifically, we demonstrated that USP7 promotes deubiquitination and stabilization of PHF8, leading to the upregulation of a group of genes, including cyclin A2, that are critical for cell growth and proliferation. The USP7-encoding gene was also transcriptionally regulated by PHF8, via positive feedback. USP7 was overexpressed in breast carcinomas, and the level of expression positively correlated with expression of PHF8 and cyclin A2 and with the histological grade of breast cancer. We showed that USP7 promotes breast carcinogenesis by stabilizing PHF8 and upregulating cyclin A2 and that the interaction between USP7 and PHF8 is augmented during DNA damage. Moreover, USP7-promoted PHF8 stabilization conferred cellular resistance to genotoxic insults and was required for the recruitment of BLM and KU70, which are both essential for DNA double-strand break repair. Our study mechanistically links USP7 to epigenetic regulation and DNA repair. Moreover, these data support the pursuit of USP7 and PHF8 as potential targets for breast cancer intervention, especially in combination with chemo- or radiotherapies.
Authors
Qian Wang, Shuai Ma, Nan Song, Xin Li, Ling Liu, Shangda Yang, Xiang Ding, Lin Shan, Xing Zhou, Dongxue Su, Yue Wang, Qi Zhang, Xinhua Liu, Na Yu, Kai Zhang, Yongfeng Shang, Zhi Yao, Lei Shi

Abstract
Homocystinuria, which typically results from cystathionine β-synthase (CBS) deficiency, is the most common defect of sulfur amino acid metabolism. CBS condenses homocysteine and serine to cystathionine that is then converted to cysteine. Individuals with homocystinuria have markedly elevated plasma levels of homocysteine and methionine and reduced concentrations of cystathionine and cysteine. Clinical disease manifestations include thromboembolism and neuropsychiatric, ocular, and skeletal complications. Here, we have shown that administration of PEGylated CBS into the circulation of homocystinuria model mice alters the extra- and intracellular equilibrium of sulfur amino acids, resulting in a decrease of approximately 75% in plasma total homocysteine (tHcy) and normalization of cysteine concentrations. Moreover, the decrease in homocysteine and the normalization of cysteine in PEGylated CBS–treated model mice were accompanied by improvement of histopathological liver symptoms and increased survival. Together, these data suggest that CBS enzyme replacement therapy (ERT) is a promising approach for the treatment of homocystinuria and that ERT for metabolic diseases may not necessitate introduction of the deficient enzyme into its natural intracellular compartment.
Authors
Erez M. Bublil, Tomas Majtan, Insun Park, Richard S. Carrillo, Helena Hůlková, Jakub Krijt, Viktor Kožich, Jan P. Kraus

Abstract
Systemic lupus erythematosus (SLE) is a devastating multisystemic autoimmune disorder. However, the molecular mechanisms underlying its pathogenesis remain elusive. Some patients with Noonan syndrome, a congenital disorder predominantly caused by gain-of-function mutations in the protein tyrosine phosphatase SH2 domain–containing PTP (SHP2), have been shown to develop SLE, suggesting a functional correlation between phosphatase activity and systemic autoimmunity. To test this directly, we measured SHP2 activity in spleen lysates isolated from lupus-prone MRL/
Authors
Jianxun Wang, Masayuki Mizui, Li-Fan Zeng, Roderick Bronson, Michele Finnell, Cox Terhorst, Vasileios C. Kyttaris, George C. Tsokos, Zhong-Yin Zhang, Maria I. Kontaridis

Abstract
Vα24-invariant natural killer T cells (NKTs) localize to tumors and have inherent antitumor properties, making them attractive chimeric antigen receptor (CAR) carriers for redirected cancer immunotherapy. However, clinical application of CAR-NKTs has been impeded, as mechanisms responsible for NKT expansion and the in vivo persistence of these cells are unknown. Here, we demonstrated that antigen-induced expansion of primary NKTs in vitro associates with the accumulation of a CD62L+ subset and exhaustion of CD62L– cells. Only CD62L+ NKTs survived and proliferated in response to secondary stimulation. When transferred to immune-deficient NSG mice, CD62L+ NKTs persisted 5 times longer than CD62L– NKTs. Moreover, CD62L+ cells transduced with a CD19-specific CAR achieved sustained tumor regression in a B cell lymphoma model. Proliferating CD62L+ cells downregulated or maintained CD62L expression when activated via T cell receptor alone or in combination with costimulatory receptors. We generated HLAnull K562 cell clones that were engineered to express CD1d and costimulatory ligands. Clone B-8-2 (HLAnullCD1dmedCD86high4-1BBLmedOX40Lhigh) induced the highest rates of NKT expansion and CD62L expression. B-8-2–expanded CAR-NKTs exhibited prolonged in vivo persistence and superior therapeutic activities in models of lymphoma and neuroblastoma. Therefore, we have identified CD62L as a marker of a distinct NKT subset endowed with high proliferative potential and have developed artificial antigen-presenting cells that generate CD62L-enriched NKTs for effective cancer immunotherapy.
Authors
Gengwen Tian, Amy N. Courtney, Bipulendu Jena, Andras Heczey, Daofeng Liu, Ekaterina Marinova, Linjie Guo, Xin Xu, Hiroki Torikai, Qianxing Mo, Gianpietro Dotti, Laurence J. Cooper, Leonid S. Metelitsa

Abstract
The cross-reactivity of T cells with pathogen- and self-derived peptides has been implicated as a pathway involved in the development of autoimmunity. However, the mechanisms that allow the clonal T cell antigen receptor (TCR) to functionally engage multiple peptide–major histocompatibility complexes (pMHC) are unclear. Here, we studied multiligand discrimination by a human, preproinsulin reactive, MHC class-I–restricted CD8+ T cell clone (1E6) that can recognize over 1 million different peptides. We generated high-resolution structures of the 1E6 TCR bound to 7 altered peptide ligands, including a pathogen-derived peptide that was an order of magnitude more potent than the natural self-peptide. Evaluation of these structures demonstrated that binding was stabilized through a conserved lock-and-key–like minimal binding footprint that enables 1E6 TCR to tolerate vast numbers of substitutions outside of this so-called hotspot. Highly potent antigens of the 1E6 TCR engaged with a strong antipathogen-like binding affinity; this engagement was governed though an energetic switch from an enthalpically to entropically driven interaction compared with the natural autoimmune ligand. Together, these data highlight how T cell cross-reactivity with pathogen-derived antigens might break self-tolerance to induce autoimmune disease.
Authors
David K. Cole, Anna M. Bulek, Garry Dolton, Andrea J. Schauenberg, Barbara Szomolay, William Rittase, Andrew Trimby, Prithiviraj Jothikumar, Anna Fuller, Ania Skowera, Jamie Rossjohn, Cheng Zhu, John J. Miles, Mark Peakman, Linda Wooldridge, Pierre J. Rizkallah, Andrew K. Sewell
No posts were found with this tag.



Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)