Advertisement
Research ArticleCell biologyOncology
Open Access | ![]() 10.1172/JCI192705
10.1172/JCI192705
SH3BP5L triggers the RAB11A-regulated integrin recycling network implicated in breast cancer metastasis
Huayi Li,1 Maria Chiara De Santis,1 Francesco A. Tucci,2,3 Daniela Tosoni,2,3 Ping Zhang,1 Meredith L. Jenkins,4 Giulia Villari,5,6 Maria Grazia Filippone,2 Elisa Guerrera,2 Simone Tealdi,7,8 Luca Gozzelino,1,8 Federico Gulluni,1 Lorenzo Prever,1 Cristina Zanini,1 Marco Forni,1 Irene Franco,9 Miriam Martini,1 John E. Burke,4,10 Guido Serini,5,6 Carlo Cosimo Campa,6,8 Salvatore Pece,2,3 Jean Piero Margaria,1,11 and Emilio Hirsch1
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Li, H. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by De Santis, M. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Tucci, F. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Tosoni, D. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Zhang, P. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Jenkins, M. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Villari, G. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Filippone, M. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Guerrera, E. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Tealdi, S. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Gozzelino, L. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Gulluni, F. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Prever, L. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Zanini, C. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Forni, M. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Franco, I. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by
Martini, M.
in:
PubMed
|
Google Scholar
|

1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Burke, J. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by
Serini, G.
in:
PubMed
|
Google Scholar
|
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Campa, C. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Pece, S. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Margaria, J. in: PubMed | Google Scholar
1Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center “G. Tarone,” University of Torino, Torino, Italy.
2IEO, European Institute of Oncology IRCCS, Milan, Italy.
3Department of Oncology and Hemato-Oncology, University of Milano, Milano, Italy.
4Department of Biochemistry and Microbiology, University of Victoria, Victoria, British Columbia, Canada.
5Department of Oncology, University of Torino Medical School, Candiolo, Torino, Italy.
6Candiolo Cancer Institute, Fondazione del Piemonte per l’Oncologia (FPO) – IRCCS, Candiolo, Torino, Italy.
7Department of Mechanical and Aerospace Engineering, Politecnico di Torino, Torino, Italy.
8Italian Institute for Genomic Medicine, Candiolo, Torino, Italy.
9Università Vita-Salute San Raffaele Milan, Italy.
10Department of Biochemistry and Molecular Biology, The University of British Columbia, Vancouver, British Columbia, Canada.
11Somatic mutation mechanisms Unit, Division of Genetics and Cellular Biology, Ospedale San Raffaele – IRCCS, Milan, Italy.
Address correspondence to: Jean Piero Margaria, Somatic Mutation Mechanisms Unit, Division of Genetics and Cell Biology, IRCCS San Raffaele Hospital, Via Olgettina 58, 20132 Milan, Italy. Phone: 0039.02.2643.2357; Email: margaria.jean@hsr.it. Or to: Emilio Hirsch, Department of Molecular Biotechnology and Health Sciences, Molecular Biotechnology Center (MBC), University of Torino, Via Nizza 52, 10126 Torino, Italy. Phone: 0039.011.670.6425; Email: emilio.hirsch@unito.it.
Authorship note: HL, MCDS, FAT, DT, and PZ contributed equally to this work and have been designated as co–first authors. CCC, SP, JPM, and EH contributed equally to this work as co–last authors.
Find articles by Hirsch, E. in: PubMed | Google Scholar
Published February 2, 2026 - More info
J Clin Invest. 2026;136(3):e192705. https://doi.org/10.1172/JCI192705.
© 2026 Li et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: February 25, 2025; Accepted: December 4, 2025

-
Results
SH3BP5L overexpression is associated with metastasis of aggressive BCs. To explore the contribution of the RAB11A-regulated vesicular recycling network in human cancers (Figure 1A), we analyzed copy number alterations (CNAs) of RAB11A, its GEFs (SH3BP5, SH3BP5L), and its GAP (TBC1D9B) across malignancies in The Cancer Genome Atlas (TCGA) database. Multiple cancer types displayed amplification of SH3BP5L, with the highest frequency (10%) in BC (Figure 1B). We therefore investigated the implications of SH3BP5L expression in this context.
 Figure 1
Figure 1SH3BP5L expression in BC and association with patient outcomes. (A) Schematic representation of the RAB11 regulatory network. RAB11-GDP is activated by its GEFs, SH3BP5L and SH3BP5, and inactivated by its GAP, TBC1D9B. (B) Copy number alterations of SH3BP5L, SH3BP5, TBC1D9B, and RAB11A across cancer types in TCGA database. Adren., adrenal gland; Oesop., esophagus; Endom., endometrium; Stoma., stomach; Pancr., pancreas; UAT, upper aerodigestive tract; Urina., urinary tract; H&L, hematopoietic and lymphoid; Intes., large intestinal; Soft t., soft tissue. (C) Violin plot showing SH3BP5L mRNA expression across tumor grades in the METABRIC dataset (n = 1,893). **P = 0.002 and ***P < 0.001, by Wilcoxon–Mann-Whitney U test. (D and E) Association of SH3BP5L expression with RFS in a composite BC cohort. (D) Continuous analysis of the log relative hazard against SH3BP5L expression. Both a linear fit (red) and a penalized spline fit (gray) are shown. The HR and log-rank test P value refer to the linear term. Shaded areas represent 95% CIs. Dashed line indicates the upper tertile cutoff. (E) Kaplan-Meier analysis of patients stratified according to SH3BP5Lhi (upper tertile) and SH3BP5Llo expression. P denotes the log-rank P value; n = 4,005. (F) Representative immunoblots of SH3BP5L and SH3BP5 expression normalized to vinculin in luminal and TNBC cell lines. (G) Immunohistochemical assessment of SH3BP5L expression in patients with BC (n = 1590). Representative images of SH3BP5Llo tumors (n = 1,470, score 0/1) and SH3BP5Lhi tumors (n = 120, score 2/3). Scale bars: 100 μm. (H) DMFS analysis of patients with SH3BP5Lhi versus SH3BP5Llo BC. P denotes the log-rank test P value; n = 1,590.
Transcriptomics analyses of public BC datasets showed that SH3BP5L mRNA levels were higher in high-grade tumors (Figure 1C) and enriched in TNBC compared with luminal tumors (Supplemental Figure 1, A and B; supplemental material available online with this article; https://doi.org/10.1172/JCI192705DS1) (26, 27). In METABRIC (26), SH3BP5L was also elevated in human epidermal growth factor receptor 2–positive (HER2+) tumors, with no significant difference between TNBC and HER2+ BC (Supplemental Figure 1B). To assess the clinical significance, we analyzed a Composite Breast Cancer cohort integrating the METABRIC and KMplotter microarray datasets to increase statistical power and avoid single-cohort biases. Both cohorts have long-term follow-up (median of 13.0 and 6.9 years for METABRIC and KMplotter, respectively) and showed a similar distribution of PAM50 subtypes (Supplemental Figure 1C). Across these datasets, SH3BP5L mRNA levels were associated with shorter relapse-free survival (RFS), with increasing expression corresponding to a monotonic, approximately linear rise in relative hazard in the aggregated cohort (Figure 1D) and in each dataset considered separately, with similar effect size and statistical significance (Supplemental Figure 1, D and E). Consistently, when patients were dichotomized, RFS was shorter for patients with SH3BP5Lhi tumors than for those with SH3BP5Llo tumors (Figure 1E).
By contrast, the close paralog SH3BP5 showed no enrichment in any molecular subtype (Supplemental Figure 1F and Supplemental Methods) and was instead associated with a favorable prognosis (Supplemental Figure 1, G and H). Protein analyses confirmed this divergence: TNBC cell lines expressed significantly more SH3BP5L than did luminal cell lines, with the highest levels detected in metastatic HCC1806 and MDA-MB-231 cells, whereas SH3BP5 showed no consistent enrichment (Figure 1F and Supplemental Figure 1I).
To confirm the clinical relevance, we assessed SH3BP5L protein expression in a retrospective BC cohort of 2,453 patients with complete clinicopathological annotation and long-term follow-up (median 14.5 years) (28, 29). A total of 1,590 viable tumor cores were scored for overall SH3BP5L staining intensity by IHC. On the basis of this evaluation, most patients (n = 1,470) were classified as SH3BP5Llo, whereas 120 were SH3BP5Lhi (Figure 1G and Table 1). SH3BP5Lhi status was associated with indicators of aggressive disease, including higher Ki67 levels, grade, and pathological tumor stage (Table 1). Clinically, SH3BP5Lhi patients showed significantly reduced distant metastasis–free survival (DMFS) (Figure 1H).
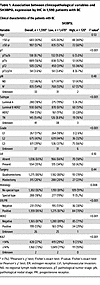 Table 1
Table 1Association between clinicopathological variables and SH3BP5L expression by IHC in 1,590 patients with BC
We then analyzed SH3BP5L expression across BC molecular subtypes. SH3BP5Lhi tumors were enriched in TNBC (16%) compared with SH3BP5Llo tumors (8.9%) or the cohort average (9.4%), and also included a higher proportion of HER2+ cases (28%) (Table 1). Patients with SH3BP5Lhi cancer had largely invasive ductal/no special type and showed no association with rarer histological variants (Table 1). Subtype-specific survival analyses revealed that high SH3BP5L expression was significantly associated with reduced DMFS in HER2+ cancers. A similar trend was observed in TNBC, although statistical significance was likely limited by the smaller number of TNBC cases (Supplemental Figure 2A). By contrast, we observed no prognostic association in luminal tumors.
In agreement, subgroup analysis in the Composite Breast Cancer cohort confirmed SH3BP5L risk stratification in HER2+ and basal-like subtypes (Supplemental Figure 2B). Conversely, SH3BP5 did not consistently stratify HER2+ and was positively associated with a good prognosis in luminal and basal-like disease (Supplemental Figure 2C).
Since HER2+ cancers already benefit from effective targeted therapies, we concentrated subsequent analyses on TNBC, for which therapeutic options remain limited and new approaches are urgently needed to reduce metastatic burden.
The GEF activity of SH3BP5L promotes TNBC cell migration via RAB11A activation. We investigated the role of SH3BP5L in aggressive BC by knockdown (KD) of SH3BP5L in MDA-MB-231 cells, a cellular model of TNBC with high SH3BP5L expression. A previously developed Förster resonance energy transfer (FRET) sensor to measure RAB11A activation (AS-RAB11A) (30, 31) highlighted a decrease in RAB11A activation upon SH3BP5L KD (siSH3BP5L) (Figure 2, A–C, and Supplemental Figure 3A). This finding was independently validated using a pull-down assay with the recombinant RAB11-GTP–interacting domain of FIP3 as a probe (30, 32). This second experiment confirmed reduced RAB11A activity with decreased SH3BP5L expression (Figure 2D, Supplemental Figure 3B, and Supplemental Methods). In contrast, overexpression of WT SH3BP5L restored and enhanced RAB11A activity, as measured by the FRET sensor (Supplemental Figure 3, C and D).
 Figure 2
Figure 2SH3BP5L affects RAB11A activation. (A) Representative pseudocolored confocal image of the FRET ratio in MDA-MB-231 cells transfected with SH3BP5L-silenced AS-RAB11A. Scale bars: 5 μm. (B) Spectrophotometry assay quantification of FRET ratio emission intensity in lysate obtained from HEK293T cells transfected with AS-RAB11A FRET sensor and SH3BP5L siRNAs. n ≥6 independent experiments. (C) Quantification of the FRET ratio from confocal image analysis of MDA-MB-231 cells transfected with AS-RAB11A sensor and SH3BP5L siRNAs. n ≥9 cells. (D) Representative Western blot of endogenous RAB11-GTP content measured with a RAB11 activation pull-down assay using GST-RAB11FIP3 (specifically binds to the active GTP loaded form of RAB11A) in SH3BP5L-depleted MDA-MB-231 cells. (E) Table representing SH3BP5L conserved regions for the binding of RAB11A and the overlapping putative SH3BP5L amino acid residues. (F) Spectrophotometry assay quantification of FRET ratio emission intensity in lysate obtained from HEK293T cells transfected with AS-RAB11A FRET sensor and mCherry-SH3BP5L(WT), mCherry-SH3BP5L(AAA), or mCherry-SH3BP5L(AK) constructs. n ≥7 independent experiments. (G) Quantification of the FRET ratio from confocal image analysis of MDA-MB-231 cells transfected with AS-RAB11A sensor and mCherry-SH3BP5L(WT), mCherry-SH3BP5L(AAA), or mCherry-SH3BP5L(AK) constructs. n ≥10 cells. (H) Representative Western blot of endogenous RAB11-GTP content measured with a RAB11 activation pull-down assay using GST-RAB11FIP3 (specifically binds to the active GTP-loaded form of RAB11A) in MDA-MB-231 cells transfected with mCherry (Ctrl), mCherry-SH3BP5L(WT), mCherry-SH3BP5L(AAA), or mCherry-SH3BP5L(AK) constructs. (I and J) Representative crystal violet staining (I) and relative quantification (J) of a Transwell assay of MDA-MB-231 cells depleted of SH3BP5L (siSH3BP5L) and transfected with mCherry-SH3BP5L(WT), mCherry-SH3BP5L(AAA), mCherry-SH3BP5L(AK), or constitutively active GFP-RAB11A(Q70L) constructs. Transfection efficiency was confirmed separately, with at least 70% of cells fluorescently labeled. Scale bars: 60 μm (I). Data represent the mean of at least 3 independent experiments ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.005, by 1-way ANOVA. Ctrl, control.
Next, we assessed the GEF activity of SH3BP5L. We introduced point mutations into conserved residues of the SH3BP5L GEF domain, previously shown to abolish SH3BP5 GEF activity (13), thereby generating the SH3BP5L(AAA) and SH3BP5L(AK) variants (Figure 2E). Measurements of RAB11A activity by spectrofluorimetric analysis, confocal microscopy, and FIP3 pull-down assays showed that SH3BP5L(AAA) and SH3BP5L(AK) induced a 50% reduction in RAB11A activation compared with SH3BP5L(WT) (Figure 2, F–H, Supplemental Figure 3E, and Supplemental Methods).
Given the involvement of SH3BP5L in BC aggressiveness (Figure 1), we hypothesized that the SH3BP5L/RAB11A axis could positively influence metastatic spreading. We induced overexpression of RAB11A(WT) and the constitutively active RAB11A(Q70L) in the TNBC cell line MDA-MB-231. Both transfections increased migration, whereas the inactive mutant RAB11A(S25N) had the opposite effect, i.e., a reduction in migration (Supplemental Figure 3, F and G). We also observed a significant reduction in migration in SH3BP5L-KD cells (siSH3BP5L) (Figure 2, I and J). Remarkably, this effect appeared specific to cell movement, as cell proliferation remained unaffected in siSH3BP5L cells (Supplemental Figure 3H). The reduction in migration was fully rescued by reintroducing WT SH3BP5L, whereas GEF-deficient mutants (AAA and AK) failed to restore the impaired migration in siSH3BP5L cells (Figure 2, I and J). Similarly, the reduced migration observed in siSH3BP5L cells was reversed by genetic activation of the cells’ downstream effector RAB11A, achieved by transfection of the constitutively active RAB11A(Q70L) mutant (Figure 2, I and J). This rescue did not change when we induced concomitantly expression of RAB11AQ70L and either one of the GEF-deficient SH3BP5L mutants in siSH3BP5L cells (Supplemental Figure 3, F and G). Notably, the overexpression of SH3BP5L, but not of its closely related homolog SH3BP5, resulted in migratory improvements (Supplemental Figure 3, I and J), pointing to a specific requirement of the GEF activity of SH3BP5L for TNBC cell migration.
Altogether, these results establish SH3BP5L as a key regulator of the migratory capacity of MDA-MB-231 cells, and this function was strictly mediated by SH3BP5L GEF activity toward RAB11A.
SH3BP5L-activated RAB11A interacts with KIF5B to direct vesicles to the plasma membrane. By activating RAB11A in TNBC cells, SH3BP5L may promote the recycling of cargo vesicles. This process is enabled by various molecular players that facilitate vesicle movement and positioning. In particular, serum stimulation can trigger cargo vesicles to recycle back to the plasma membrane and facilitate cell migration by providing multiple cues, including growth factors and extracellular matrix (ECM) components. To pinpoint specific molecular actors that interact with SH3BP5L and contribute to its GEF activity–dependent role in metastasis, we performed proteomics analysis of SH3BP5L-GFP pull-down assays. To this aim, we conducted a comparative analysis of the protein complexes immunoprecipitated with SH3BP5L in 2 distinct cellular scenarios: (a) a control condition, represented by cells maintained under serum starvation (serum-free) and (b) a test condition, represented by cells subjected to serum starvation followed by a 40-minute reexposure to an FBS treatment (serum-fed) leading to plasma membrane recycling of cargo proteins (32–35) (Figure 3A). This approach revealed substantial enrichment of vesicular transport proteins in the serum-treated SH3BP5L sample (Supplemental Figure 4A).
 Figure 3
Figure 3SH3BP5L interacts with KIF5B to recycle cargo back to the plasma membrane. (A) Volcano plot of mass spectrometry analysis performed from GFP immunoprecipitation of HEK293T cells transfected with GFP or GFP-SH3BP5L in the control (serum-free) or recycling (serum-fed) condition. (B and C) Bubble plot illustrating the relationship between the average expression and Top3 metric for (B) RAB and (C) kinesin family proteins. Bubble size represents the log fold change (log FC), and bubble color corresponds to the P value. (D) Representative Western blot of GFP immunoprecipitation performed in HEK293T cells transfected with GFP, GFP-SH3BP5L(WT), GFP-SH3BP5L(AAA) , or GFP-SH3BP5L(AK) constructs in serum-starved or -fed conditions. (E and F) Representative confocal images (E) and quantification (F) of a PLA for RAB11A and KIF5B in MDA-MB-231 cells. n = 6 images for each group from 3 independent experiments. Scale bars: 5 μm. (G and H) Representative Western blots (G) and relative quantification (H) of GFP immunoprecipitation performed on HEK293T cells transfected with GFP, GFP-RAB11A(WT), GFP-RAB11AQ70L, or GFP-RAB11A(S25N) constructs. (I and J) Representative Western blots (I) and relative quantification (J) of a pull-down assay for GST-RAB11A loaded with GDP or GTPγS performed on HEK293T cells transfected with HA-KIF5B. (K) Kymograph of fluorescence intensity for mCherry-SH3BP5L (red) and GFP-KIF5B (green) along an iRFP-RAB11A-labeled (magenta) vesicle over time. Merge shows the colocalization dynamics. (L) Representative image of the vesicle positive for iRFP-RAB11A, mCherry-SH3BP5L, and GFP-KIF5B (top) showing its transport toward the plasma membrane (PM). Quantification of relative fluorescence intensity over time for iRFP-RAB11A, mCherry-SH3BP5L, and GFP-KIF5B, with shaded regions indicating standard deviation (bottom). Scale bars: 1 μm. Data represent the mean of at least 3 independent experiments ± SEM. **P < 0.01, ***P < 0.005, and ****P < 0.001, by 1-way ANOVA.
Through this procedure, we identified elements of the SH3BP5L interactome, from which we selected 92 proteins associated with intracellular vesicular traffic, including Rabs, kinesins, as well as microtubule components (Figure 3A). Statistical analysis highlighted RAB11A as the most enriched candidate among all the Rabs captured by our assay (Figure 3B), thus confirming the pivotal role of RAB11A as the primary target modulated by SH3BP5L. Particularly striking was the augmented interaction of the complex with the kinesin family member 5B (KIF5B) (Figure 3C).
Next, we validated the interaction between the SH3BP5L complex and KIF5B using immunoprecipitation. We found that the interactions between SH3BP5L, RAB11A, and KIF5B were significantly enhanced following serum stimulation and that these interactions were blocked by SH3BP5L-inactivating mutations (Figure 3D, Supplemental Figure 4, B and C, and Supplemental Methods). The serum-induced association between RAB11A and KIF5B was further confirmed through proximity ligation assays (PLAs). The assay demonstrated that these 2 proteins were assembled in a common complex (Figure 3, E and F, Supplemental Figure 4, D and E, and Supplemental Methods). Conversely, PLA analysis revealed negligible interactions between KIF5B and SH3BP5, the homolog of SH3BP5L (Supplemental Figure 4, F and G). Collectively, these findings support the assumption that SH3BP5L, rather than SH3BP5, activates a pool of RAB11A, which can recruit KIF5B, potentially coordinating the anterograde transport of metastasis-promoting cargo vesicles.
To further validate this model, we examined the interplay between RAB11A and KIF5B through anti-GFP immunoprecipitation and a PLA assay. Consistent with our hypothesis, both assays confirmed that KIF5B interacts with both WT GFP-RAB11A and its constitutively active form (Q70L), but not with the dominant-negative form (S25N) (Figure 3, G and H, Supplemental Figure 4, F and G, and Supplemental Methods). In vitro GST pull-down assays provided additional support, showing that KIF5B preferentially interacted with RAB11A in its GTP-bound form (GST-RAB11AGTPγS) over the GDP-bound form (GST-RAB11AGDP) (Figure 3, I and J, and Supplemental Methods). Analysis of the interaction between GTP-loaded RAB11A and a KIF5B dimer by AlphaFold3 (36) showed a potential direct interaction between these proteins at the C-terminal end of KIF5B (Supplemental Figure 4H), with preferential affinity for GTP- versus GDP-loaded RAB11 (Supplemental Figure 4I).
Next, to elucidate the dynamics of SH3BP5L, RAB11A, and KIF5B interactions, we performed kymograph tracking analysis of cells transfected with mCherry-SH3BP5L, iRFP-RAB11A, and GFP-KIF5B using confocal microscopy. This analysis revealed the enrichment of SH3BP5L followed by the recruitment of KIF5B to nascent RAB11A+ vesicles. These vesicles emerged from the perinucleus-positioned organelles and were directed toward the plasma membrane (Figure 3, K and L, and Supplemental Methods). Together, these data indicate that SH3BP5L activated RAB11A, which in turn promoted the recruitment of the anterograde motor protein KIF5B, suggesting a role for SH3BP5L in coordinating cargo vesicle recycling to the plasma membrane.
SH3BP5L overexpression boosts plasma membrane–directed recycling of active integrin β1 in TNBC cells. To assess the function of SH3BP5L in activating cargo recycling, we used a high-content imaging (HCI) pipeline. This in silico method infers 3D subcellular distribution by combining fluorescence prediction and segmentation (37, 38) (Supplemental Figure 5A). The prediction utilizes a neural network trained to recognize subcellular compartments in bright-field images, without the need for antibody labeling of each compartment. This approach circumvented the technical challenges of spectral separation in multicolor fluorescence imaging. The published training protocol was applied to mesenchymal MDA-MB-231 cells as well as to more basal-like MDA-MB-468 cells to recognize subcellular membranes associated with Golgi (RCAS1), the lysosome (LAMP1), recycling compartments (RAB11A), mitochondria (MitoTracker dye), and the plasma membrane (Cell Mask dye). After verifying the accuracy of the prediction (Supplemental Figure 5B), we used this method to compare the subcellular localization of SH3BP5L and its paralog SH3BP5 in MDA-MB-231 cells. Both proteins were enriched at the plasma membrane, consistent with their role as activators of RAB11-dependent anterograde transport. However, SH3BP5 showed stronger localization to mitochondria, whereas SH3BP5L was more enriched in the perinuclear RAB11+ recycling compartment (Supplemental Figure 5C). These differences point to functional specialization between the 2 paralogs.
Next, we used the HCI pipeline to functionally test if loss of SH3BP5L affects the RAB11-dependent recycling of adhesion receptors involved in metastasis. For example, BC cell motility relies on the efficient endocytosis and recycling of integrin heterodimers, which form a recycling loop between the endosomal compartment and the plasma membrane. The endosomal compartment processes endocytosed cargos from the cell surface, directing them either for degradation or recycling to the plasma membrane (39, 40). Similarly, other key adhesion receptors like CD44 control metastasis (41) and require RAB11 to recycle back once endocytosed (42). Therefore, we focused our attention on CD44 and integrin subunits such as β1, α2, α3, α4, α5, αV, and α6. SH3BP5L KD led to a selective reduction of integrin β1 (ITGB1) and integrin α3 (ITGA3) at the plasma membrane, whereas the localization of other integrins and CD44 remained unaffected (Figure 4A and Supplemental Methods). To further validate the accuracy of this approach, the decrease in plasma membrane content of ITGB1 and ITGA3 in cells where SH3BP5L was downregulated was confirmed by FACS staining (Figure 4, B and C). These findings demonstrate that SH3BP5L selectively regulated β1 and α3 integrin recycling and confirm the ability of the HCI pipeline to faithfully capture the intracellular distribution of ITGB1 in BC cells.
 Figure 4
Figure 4SH3BP5L boosts ITGB1 recycling. (A) AI-driven quantification of different integrin subunits and CD44 at the plasma membrane of MDA-MB-231 cells upon SH3BP5L depletion. n ≥12 cells from 3 independent experiments. (B and C) FACS quantification of plasma membrane ITGB1 (B) and ITGA3 (C) in MDA-MB-231 cells upon SH3BP5L depletion. (D) AI-driven quantification of active ITGB1 (with mAb 9EG7) subcellular distribution in MCF7 cells overexpressing SH3BP5L(WT). n = 22 control cells, n = 18 SH3BP5L(WT) cells. (E) AI-driven quantification of active ITGB1 (mAb 9EG7) subcellular distribution in MDA-MB-231 cells depleted of SH3BP5L. n = 43 control cells, n = 37 siSH3BP5L cells. (F and G) FACS quantification of active ITGB1 (mAb 9EG7) recycling at 30 minutes in MDA-MB-231 cells transfected with 2 different SH3BP5L siRNAs or (F) with mCherry, mCherry-SH3BP5L(WT), mCherry-SH3BP5L(AAA), or mCherry-SH3BP5L(AK) constructs (G). (H and I) Real-time adhesion assay of MDA-MB-231 cells transfected with control or SH3BP5L siRNA and seeded on laminin (H) or fibronectin (I). Data represent the mean of at least 3 independent experiments ± SEM. *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.001, by 1- or 2-way ANOVA followed by Bonferroni post hoc test as appropriate.
We next examined whether SH3BP5L regulates recycling of the active conformation of β1 integrin, as only the ligand-binding active pool engages the extracellular matrix and transduces signals required for migration (43). To validate the ability of the HCI pipeline to detect active ITGB1, fixed MDA-MB-231 cells were labeled with antibodies recognizing active (9EG7), inactive (mAb13), or total ITGB1 (P5D2) (44). Consistent with previous reports (43, 45), total ITGB1 in control cells was primarily localized to the plasma membrane, while active ITGB1 was also present in intracellular compartments (Supplemental Figure 5D). The unrelated cytoplasmic protein mCherry showed no specific compartmentalization (Supplemental Figure 5D). With this validation of the HCI pipeline, we further investigated whether induced overexpression of SH3BP5L alters active ITGB1 distribution in MCF7 cells, a nonmetastatic luminal BC cell line that expresses physiological levels of this GEF (Figure 1F). In these cells, SH3BP5L overexpression increased active ITGB1 in the RAB11A+ compartment and at the plasma membrane, while reducing its accumulation in lysosomes as indicated by fewer LAMP1+ vesicles (Figure 4D and Supplemental Methods).
Conversely, SH3BP5L KD in the TNBC cell lines MDA-MB-231 and MDA-MB-468 reduced active ITGB1 levels in the RAB11A+ compartment and at the plasma membrane, while promoting its accumulation in LAMP1+ lysosomes (Figure 4E and Supplemental Figure 5E). These results indicate that SH3BP5L promotes plasma membrane delivery of active ITGB1 by controlling its intracellular trafficking. To directly assess recycling dynamics, we performed antibody-based assays using 9EG7 and measured surface reexposure of active ITGB1 by flow cytometry (43). SH3BP5L depletion markedly impaired active ITGB1 recycling in 2 TNBC cell lines (Figure 4F, Supplemental Figure 5, F and G, and Supplemental Figure 6A). We observed a similar defect when the GEF activity of SH3BP5L was abolished using catalytically inactive mutants (Figure 4G and Supplemental Figure 6B).
Finally, to evaluate the functional consequences of this defect, we examined cell adhesion to specific ECM ligands. SH3BP5LKD in MDA-MB-231 cells significantly reduced adhesion to laminin, a known ligand for α3β1 activation, but had no effect on fibronectin, which primarily engages other β1-containing integrin heterodimers (Figure 4, H and I). Together, these results demonstrate that SH3BP5L controlled the recycling of α3β1 integrin in a GEF-dependent manner, providing a mechanistic link to its role in sustaining TNBC cell adhesion and metastatic potential.
SH3BP5L couples RAB11A activation to TNBC invasion in fish, mouse, and patient-derived models. We next sought to investigate the role of SH3BP5L in TNBC metastasis in vivo. First, we generated a SH3BP5L-KO (koSH3BP5L) MDA-MB-231 cell line using CRISPR/Cas9, along with a control cell line, in which SH3BP5L expression was rescued by expressing WT SH3BP5L (Supplemental Figure 7A). As previously observed (Supplemental Figure 3H), reduction of SH3BP5L expression did not affect cell proliferation but significantly decreased cell migration in vitro (Figure 5A, Supplemental Figure 7, B–D, and Supplemental Methods). We then injected these engineered KO cells into the tail vein of NSG mice. Five weeks after injection, koSH3BP5L cells exhibited a marked reduction in lung metastasis compared with WT cells or koSH3BP5L cells rescued by mCherry-SH3BP5L(WT) expression (Figure 5, B and C). We repeated the experiment in a zebrafish embryo model (46–48), which allowed us to perform live imaging of cancer cell dissemination. In agreement with our observations in mice, koSH3BP5L MDA-MB-231 and MDA-MB-468 cells exhibited a significantly reduced incidence of metastasis and fewer disseminated foci in zebrafish tails compared with WT cells. Reintroduction of SH3BP5L expression in koSH3BP5L cells restored metastatic spreading to levels comparable to those of wild-type controls (Figure 5, D and E, and Supplemental Figure 7, E and F).
 Figure 5
Figure 5SH3BP5L enhances RAB11A-mediated invasion and metastasis. (A) Representative image of Transwell (upper) and single-cell tracking (bottom) assay of MDA-MB-231 cells KO for SH3BP5L cells and transfected with mCherry-SH3BP5L(WT) (koSH3BP5L/mCherry-SH3BP5L(WT)). For cell tracking, n = 25 (Ctrl), n = 36 (koSH3BP5L), n = 21 (koSH3BP5L/mCherry-SH3BP5L(WT)) cells, respectively. Scale bars: 600 μm and 300 μm (enlarged insets). (B and C) Representative images (B) and relative quantification (C) of H&E staining of lung metastases in NSG mice injected in the tail vein with koSH3BP5L in MDA-MB-231 cells and transfected with mCherry-SH3BP5L(WT) (koSH3BP5L/mCherry-SH3BP5L(WT)). Black arrowheads indicate metastatic foci. Each dot in the graph in C is representative of an injected mouse (n = 5). Scale bars: 60 μm. (D and E) Representative images (D) and relative quantification (E) of metastasized foci in zebrafish injected with MDA-MB-231 cells with koSH3BP5L and transfected with mCherry-SH3BP5L(WT) (koSH3BP5L/SH3BP5L(WT)), mCherry-SH3BP5L(AAA) (koSH3BP5L/SH3BP5L(AAA)), mCherry-SH3BP5L(AK) (koSH3BP5L/SH3BP5LAK), or GFP-RAB11A(Q70L) (koSH3BP5L/RAB11AQ70L). Arrowheads indicate metastatic foci in zebrafish tails. Each dot in the graph in E is representative of an injected zebrafish (n ≥30). Scale bars: 0.5 mm. (F and G) Representative images (F) and relative quantification (G) of H&E staining of lung metastases in NSG mice orthotopically injected with MDA-MB-231 cells with koSH3BP5L and transfected with mCherry-SH3BP5L(WT) (koSH3BP5L/SH3BP5L(WT)), mCherry-SH3BP5L(AAA) (koSH3BP5L/SH3BP5L(AAA)), mCherry-SH3BP5L(AK) (koSH3BP5L/SH3BP5L(AK)), or GFP-RAB11A(Q70L) (koSH3BP5L/RAB11A(Q70L)). Each dot in the graph in G is representative of an injected mouse (n ≥3). Scale bars: 600 μm and 300 μm (enlarged insets). (H and I) In vitro Matrigel Transwell invasion assay (left) and spheroid-forming assay (right) of cells derived from 2 patients with TNBC (patient 1, H; patient 2, I) transduced with either control or 2 independent lentiviral shRNAs targeting SH3BP5L (left; n = 12 fields per group from 3 independent experiments; right: n = 8). Data represent the mean of at least 3 independent experiments ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.005, by 1-way ANOVA.
Next, we performed rescue experiments using WT and mutant forms of SH3BP5L, as well as WT and constitutively active RAB11A, to determine whether SH3BP5L GEF activity influences in vivo invasion. We injected koSH3BP5L MDA-MB-231 cells into the yolk sac of zebrafish larvae and quantified their spread to the tail region. Consistent with the Transwell assay results (Supplemental Figure 3, F and G), we observed reduced cell spreading in larvae injected with koSH3BP5L cells and rescue of this defect upon introduction of an active RAB11A(Q70L) mutant into the KO cells (Figure 5, D and E). In contrast, reintroducing WT, but not GEF-deficient, SH3BP5L, into koSH3BP5L cells restored distant spreading. These experiments demonstrate that cell dissemination was dependent on SH3BP5L GEF activity toward RAB11A (Figure 5, D and E). We observed similar results in NSG mice as, 5 weeks after injection, restoring RAB11A activity increased the number of lung metastases in SH3BP5L-KO cells, whereas impairing SH3BP5L GEF function reduced lung metastases (Figure 5, F and G).
Finally, to assess the clinical relevance of SH3BP5L expression, we used 2 TNBC patient-derived xenograft (PDX) models along with a patient-derived tumor spheroid (PDTS) assay, which is commonly used as a functional readout for tumor-initiating cell (TIC) activity in BC (49). SH3BP5L KD markedly impaired cell invasion and spheroid formation but had no effect on proliferation (Figure 5, H and I, and Supplemental Figure 7, G and H). As TIC-like activity is frequently linked to metastatic dissemination, these findings align with our in vitro and in vivo models showing that the GEF activity of SH3BP5L promoted TNBC migration, invasion, and distant colonization.






-
Discussion
The metastatic progression of BC remains a major clinical challenge, particularly in subtypes with limited therapeutic options. In this study, we identified SH3BP5L as a GEF for RAB11A that is upregulated in BCs with aggressive features. Analysis of large patient cohorts demonstrated that high SH3BP5L expression correlated with advanced tumor stage, high grade, and poor outcomes. Importantly, SH3BP5Lhi tumors were enriched not only in TNBC but also in HER2+ cancers, and in both subtypes high SH3BP5L expression was associated with reduced DMFS. These findings establish SH3BP5L as a clinically relevant marker of poor prognosis across aggressive BC subtypes. Nonetheless, our mechanistic studies focused on TNBC for 3 reasons. First, transcriptomics analyses in both METABRIC and TCGA consistently demonstrated increased SH3BP5L expression in TNBC compared with luminal tumors, and high expression was specifically linked to poor RFS in TNBC. Second, TNBC is the BC subtype most prone to early and frequent metastatic dissemination (1). Third, unlike HER2+ cancers, where multiple targeted therapies have markedly improved patient outcomes, TNBC remains without comparably effective options.
RAB11 activity assays confirmed that SH3BP5L activated RAB11A in TNBC cells, while coimmunoprecipitation assays demonstrated a specific interaction between active RAB11A and KIF5B, a kinesin motor protein essential for anterograde intracellular cargo transport. In vitro binding assays further showed that KIF5B exhibited a higher affinity for GTP-bound, rather than GDP-bound, RAB11A, supporting a model in which SH3BP5L activates RAB11A, enabling its GTP-bound form to preferentially associate with KIF5B. KIF5B had previously been identified as a RAB11 interactor through BioID analysis (24), but our data now provide direct functional evidence that SH3BP5L-mediated RAB11 activation facilitates this interaction. AlphaFold3 prediction and imaging data aligned with this model, demonstrating that SH3BP5L-mediated activation of RAB11A enabled its association with KIF5B and promoted vesicle trafficking from the perinuclear region to the plasma membrane. Consistent with this, our proteomics analysis did not detect interactors typically associated with the inactive GDP-bound state of RAB11A. In particular, KIF5A, a kinesin known to bind RAB11A-GDP (50), was absent, indicating that SH3BP5L reshaped the RAB11A interaction landscape by maintaining it in the active GTP-bound state.
Dynamic kymograph tracking further reinforced this model by revealing the temporal organization of the SH3BP5L-RAB11A-KIF5B complex in living cells. SH3BP5L promoted the formation of anterograde vesicles containing integrins, including ITGB1, and SH3BP5L KD reduced steady-state levels of active ITGB1 at the plasma membrane. This selective effect on active ITGB1 recycling was accompanied by impaired migration and spreading of SH3BP5L-deficient TNBC cells. Since ITGB1 recycling is known to facilitate cancer cell motility and invasion (51), our findings position SH3BP5L as a central regulator that couples RAB11 activation to integrin recycling through KIF5B in TNBC cells.
Mechanistically, these results converge with our integrin trafficking studies showing that SH3BP5L selectively controlled the recycling of the laminin-binding α3β1 heterodimer. Although the recycling of other integrins appeared unaffected, this specificity was sufficient to drive major changes in cell migration and metastatic dissemination. Indeed, α3β1 has been repeatedly implicated as a critical driver of invasion and poor prognosis in basal-like and TNBCs (52, 53). Its engagement with laminin, rather than with other ECM components, provides essential cues for directional motility and metastatic progression. In support of this, SH3BP5L-depleted cells exhibited impaired adhesion and spreading on laminin, but not on fibronectin, indicating that loss of α3β1 recycling selectively disabled the migratory machinery relevant to metastasis. These findings are consistent with prior reports documenting that functional blockade or genetic silencing of integrin α3 is sufficient to suppress adhesion, migration, and metastatic invasion in BC models (54–58). Together, our results and previous work highlight α3β1 as a pivotal effector whose trafficking is controlled by SH3BP5L/RAB11A signaling. Future studies will be needed to define the molecular basis of this specificity and clarify how SH3BP5L discriminates α3β1 from other β1-containing integrins during recycling.
The functional significance of this pathway was first demonstrated in vivo, where zebrafish and mouse metastasis models showed that SH3BP5L GEF activity is required for dissemination and colonization. This conclusion was further reinforced by ex vivo validation in short-term cultures derived from tumors of patients with TNBC, which preserved the heterogeneity of the parental lesions and provided a clinically relevant platform to test SH3BP5L dependency. In these patient-derived systems, SH3BP5L KD markedly impaired invasion and spheroid formation while leaving proliferation unaffected. The ability to form spheroids in nonadherent conditions is commonly used as a functional readout of tumor-initiating potential (49, 59), a property frequently associated with metastatic dissemination (60). Accordingly, the reduced spheroid formation observed after SH3BP5L silencing was consistent with the impaired invasion and colonization seen in vivo. Taken together with the retrospective cohort analysis, these findings establish SH3BP5L as both a prognostic marker and a functional driver of TNBC aggressiveness.
From a therapeutic perspective, our findings highlight SH3BP5L as an attractive target to counteract BC dissemination. Unlike direct integrin antagonists (40), which have largely failed in clinical trials due to redundancy and compensatory mechanisms at the receptor level (61), inhibition of SH3BP5L would act upstream by blocking integrin recycling, thereby indirectly reducing tumor cell adhesion and dissemination. One potential concern for this strategy is the presence of the close paralog SH3BP5. Yet our data indicate that the 2 proteins fulfilled clearly distinct functions: SH3BP5L was enriched in the perinuclear RAB11+ recycling compartment, bound KIF5B, and sustained integrin recycling and invasion, whereas SH3BP5 predominantly localized to mitochondria and lacked these activities. High SH3BP5L expression correlated with reduced survival in BC cohorts, while SH3BP5 showed the opposite trend. These spatial differences, together with the higher catalytic efficiency of SH3BP5L toward RAB11A, offer a plausible mechanistic explanation for the opposing clinical associations of the 2 paralogs. This divergence makes it unlikely that SH3BP5 could compensate for SH3BP5L inhibition, but at the same time it emphasizes the importance of achieving stringent isoform selectivity. Encouragingly, selective pharmacological targeting of GEFs is feasible, and there is increasing recognition of their druggability in different disease contexts (62). A prominent precedent is provided by brefeldin A, which selectively inhibits only a subset of the ARF1 GEF family by stabilizing an abortive ARF-GEF intermediate, while other ARF1 GEFs remain unaffected (63). Although no inhibitors of SH3BP5L currently exist, our genetic proof of concept provides a compelling rationale for developing small molecules or peptidomimetics capable of selectively blocking SH3BP5L activity or its interaction with RAB11A.
In conclusion, we identify SH3BP5L as what we believe to be the first RAB11 GEF with a defined role in BC metastasis. High SH3BP5L expression marked aggressive tumors and correlated with poor outcomes in HER2+ and TNBC. Mechanistically, SH3BP5L coupled RAB11A activation to KIF5B-mediated transport of α3β1 integrins, driving invasion and dissemination. Functional validation in cell lines, animal models, and patient-derived cultures revealed SH3BP5L as both a prognostic determinant and a therapeutic vulnerability in TNBC. Targeting SH3BP5L-dependent trafficking may thus offer a new strategy to counteract metastatic progression in a subtype of BC that urgently requires better treatment options (Figure 6).
 Figure 6
Figure 6Graphical abstract. Schematic representation illustrates that in models expressing high levels of SH3BP5L, SHBP5L interacts with activated RAB11A (RAB11-GTP), which binds KIF5B to facilitate active ITGB1 recycling, ultimately driving metastatic dissemination.




Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)