Reproductive biology

Abstract
Following migration of primordial germ cells to the genital ridge, oogonia undergo several rounds of mitotic division and enter meiosis at approximately E13.5. Most oocytes arrest in the dictyate (diplotene) stage of meiosis circa E18.5. The genes necessary to drive oocyte differentiation in parallel with meiosis are unknown. Here, we have investigated whether expression of spermatogenesis and oogenesis bHLH transcription factor 1 (
Authors
Yong-Hyun Shin, Yu Ren, Hitomi Suzuki, Kayla J. Golnoski, Hyo won Ahn, Vasil Mico, Aleksandar Rajkovic

Abstract
MicroRNAs (miRNAs) are negative modulators of gene expression that fine-tune numerous biological processes. miRNA loss-of-function rarely results in highly penetrant phenotypes, but rather, influences cellular responses to physiologic and pathophysiologic stresses. Here, we have reported that a single member of the evolutionarily conserved miR-7 family, miR-7a2, is essential for normal pituitary development and hypothalamic-pituitary-gonadal (HPG) function in adulthood. Genetic deletion of
Authors
Kashan Ahmed, Mary P. LaPierre, Emanuel Gasser, Rémy Denzler, Yinjie Yang, Thomas Rülicke, Jukka Kero, Mathieu Latreille, Markus Stoffel

Abstract
Autoimmune responses to meiotic germ cell antigens (MGCA) that are expressed on sperm and testis occur in human infertility and after vasectomy. Many MGCA are also expressed as cancer/testis antigens (CTA) in human cancers, but the tolerance status of MGCA has not been investigated. MGCA are considered to be uniformly immunogenic and nontolerogenic, and the prevailing view posits that MGCA are sequestered behind the Sertoli cell barrier in seminiferous tubules. Here, we have shown that only some murine MGCA are sequestered. Nonsequestered MCGA (NS-MGCA) egressed from normal tubules, as evidenced by their ability to interact with systemically injected antibodies and form localized immune complexes outside the Sertoli cell barrier. NS-MGCA derived from cell fragments that were discarded by spermatids during spermiation. They egressed as cargo in residual bodies and maintained Treg-dependent physiological tolerance. In contrast, sequestered MGCA (S-MGCA) were undetectable in residual bodies and were nontolerogenic. Unlike postvasectomy autoantibodies, which have been shown to mainly target S-MGCA, autoantibodies produced by normal mice with transient Treg depletion that developed autoimmune orchitis exclusively targeted NS-MGCA. We conclude that spermiation, a physiological checkpoint in spermatogenesis, determines the egress and tolerogenicity of MGCA. Our findings will affect target antigen selection in testis and sperm autoimmunity and the immune responses to CTA in male cancer patients.
Authors
Kenneth S.K. Tung, Jessica Harakal, Hui Qiao, Claudia Rival, Jonathan C.H. Li, Alberta G.A. Paul, Karen Wheeler, Patcharin Pramoonjago, Constance M. Grafer, Wei Sun, Robert D. Sampson, Elissa W.P. Wong, Prabhakara P. Reddi, Umesh S. Deshmukh, Daniel M. Hardy, Huanghui Tang, C. Yan Cheng, Erwin Goldberg
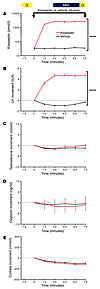
Abstract
Authors
Alexander N. Comninos, Matthew B. Wall, Lysia Demetriou, Amar J. Shah, Sophie A. Clarke, Shakunthala Narayanaswamy, Alexander Nesbitt, Chioma Izzi-Engbeaya, Julia K. Prague, Ali Abbara, Risheka Ratnasabapathy, Victoria Salem, Gurjinder M. Nijher, Channa N. Jayasena, Mark Tanner, Paul Bassett, Amrish Mehta, Eugenii A. Rabiner, Christoph Hönigsperger, Meire Ribeiro Silva, Ole Kristian Brandtzaeg, Elsa Lundanes, Steven Ray Wilson, Rachel C. Brown, Sarah A. Thomas, Stephen R. Bloom, Waljit S. Dhillo

Abstract
Authors
Hooman Mirzakhani, Augusto A. Litonjua, Thomas F. McElrath, George O’Connor, Aviva Lee-Parritz, Ronald Iverson, George Macones, Robert C. Strunk, Leonard B. Bacharier, Robert Zeiger, Bruce W. Hollis, Diane E. Handy, Amitabh Sharma, Nancy Laranjo, Vincent Carey, Weilliang Qiu, Marc Santolini, Shikang Liu, Divya Chhabra, Daniel A. Enquobahrie, Michelle A. Williams, Joseph Loscalzo, Scott T. Weiss

Abstract
Inflammation and oxidative stress are known risk factors for preterm birth (PTB); however, the mechanisms and pathways that influence this condition are not fully described. Previously, we showed that mTORC1 signaling is increased in mice harboring a uterine-specific deletion of transformation-related protein 53 (
Authors
Wenbo Deng, Jeeyeon Cha, Jia Yuan, Hirofumi Haraguchi, Amanda Bartos, Emma Leishman, Benoit Viollet, Heather B. Bradshaw, Yasushi Hirota, Sudhansu K. Dey

Abstract
Authors
Eleftheria Lefkou, Apostolos Mamopoulos, Themistoklis Dagklis, Christos Vosnakis, David Rousso, Guillermina Girardi

Abstract
Preeclampsia is a hypertensive disorder of pregnancy in which patients develop profound sensitivity to vasopressors, such as angiotensin II, and is associated with substantial morbidity for the mother and fetus. Enhanced vasoconstrictor sensitivity and elevations in soluble fms-like tyrosine kinase 1 (sFLT1), a circulating antiangiogenic protein, precede clinical signs and symptoms of preeclampsia. Here, we report that overexpression of
Authors
Suzanne D. Burke, Zsuzsanna K. Zsengellér, Eliyahu V. Khankin, Agnes S. Lo, Augustine Rajakumar, Jennifer J. DuPont, Amy McCurley, Mary E. Moss, Dongsheng Zhang, Christopher D. Clark, Alice Wang, Ellen W. Seely, Peter M. Kang, Isaac E. Stillman, Iris Z. Jaffe, S. Ananth Karumanchi

Abstract
Maternal cigarette smoking during pregnancy remains one of the most common and preventable causes of fetal growth restriction (FGR), a condition in which a fetus is unable to achieve its genetically determined potential size. Even though epidemiologic evidence clearly links maternal cigarette smoking with FGR, insight into the molecular mechanisms of cigarette smoke–induced FGR is lacking. Here, we performed transcriptional profiling of placentas obtained from smoking mothers who delivered growth-restricted infants and identified secreted frizzled-related protein 1 (sFRP1), an extracellular antagonist of endogenous WNT signaling, as a candidate molecule. sFRP1 mRNA and protein levels were markedly upregulated (~10 fold) in placentas from smoking mothers compared with those from nonsmokers. In pregnant mice, adenovirus-mediated overexpression of sFRP1 led to FGR, increased karyorrhexis in the junctional zone, and decreased proliferation of labyrinthine trophoblasts. Consistent with our hypothesis that placental WNT signaling is suppressed in maternal smokers, we found that exposure to carbon monoxide analogs led to reduced WNT signaling, increased
Authors
Alice Wang, Zsuzsanna K. Zsengellér, Jonathan L. Hecht, Roberto Buccafusca, Suzanne D. Burke, Augustine Rajakumar, Emily Weingart, Paul B. Yu, Saira Salahuddin, S. Ananth Karumanchi

Abstract
Authors
Rose G. Radin, Sunni L. Mumford, Robert M. Silver, Laurie L. Lesher, Noya Galai, David Faraggi, Jean Wactawski-Wende, Janet M. Townsend, Anne M. Lynch, Hyagriv N. Simhan, Lindsey A. Sjaarda, Neil J. Perkins, Shvetha M. Zarek, Karen C. Schliep, Enrique F. Schisterman




Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)