Advertisement
Research ArticleClinical ResearchMetabolism
Open Access | ![]() 10.1172/JCI198387
10.1172/JCI198387
Altered lipid metabolism and inflammatory programs associate with adipocyte loss in familial partial lipodystrophy 2
Jessica N. Maung,1 Rebecca L. Schill,1 Akira Nishii,1 Maria Foss de Freitas,2 Bonje N. Obua,3 Marcus Nygård,4 Maria D. Mendez-Casillas,1 Isabel D.K. Hermsmeyer,1 Donatella Gilio,2 Ozge Besci,2 Yang Chen,1 Brian Desrosiers,1 Rose E. Adler,1 Anabela D. Gomes,2 Merve Celik Guler,2 Hiroyuki Mori,1 Romina M. Uranga,1 Ziru Li,1 Hadla Hariri,1 Liping Zhang,1 Anderson de Paula Souza,2 Keegan S. Hoose,1 Kenneth T. Lewis,1 Taryn A. Hetrick,1 Paul Cederna,5 Carey N. Lumeng,1,2 Susanne Mandrup,4 Elif A. Oral,2 and Ormond A. MacDougald1,2
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by
Maung, J.
in:
PubMed
|
Google Scholar
|

1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Schill, R. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Nishii, A. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Foss de Freitas, M. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Obua, B. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Nygård, M. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Mendez-Casillas, M. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Hermsmeyer, I. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Gilio, D. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Besci, O. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Chen, Y. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Desrosiers, B. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Adler, R. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Gomes, A. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Celik Guler, M. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Mori, H. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Uranga, R. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Li, Z. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Hariri, H. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Zhang, L. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by de Paula Souza, A. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Hoose, K. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by
Lewis, K.
in:
PubMed
|
Google Scholar
|
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Hetrick, T. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by Cederna, P. in: PubMed | Google Scholar
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by
Lumeng, C.
in:
PubMed
|
Google Scholar
|
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by
Mandrup, S.
in:
PubMed
|
Google Scholar
|
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by
Oral, E.
in:
PubMed
|
Google Scholar
|
1Department of Molecular & Integrative Physiology,
2Department of Internal Medicine, and
3Cellular and Molecular Biology Program, University of Michigan Medical School, Ann Arbor, Michigan, USA.
4Center for Functional Genomics and Tissue Plasticity (ATLAS), Department of Biochemistry and Molecular Biology, University of Southern Denmark (SDU), Odense, Denmark.
5Department of Surgery, University of Michigan Medical School, Ann Arbor, Michigan, USA.
Address correspondence to: Ormond A. MacDougald, North Campus Research Complex, 2800 Plymouth Road, Building 25, Office 3686, Ann Arbor, Michigan, 48109, USA. Phone: 734.647.7721; Email: macdouga@umich.edu.
Find articles by
MacDougald, O.
in:
PubMed
|
Google Scholar
|
Published November 11, 2025 - More info
J Clin Invest. 2026;136(1):e198387. https://doi.org/10.1172/JCI198387.
© 2025, Maung et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: July 24, 2025; Accepted: November 6, 2025

-
Results
Participants with FPLD2 display loss of adipose tissue and metabolic dysfunction. To investigate how pathogenic LMNA variants drive FPLD2 progression, we recruited 8 families into the Longitudinal Evaluation of Adiposity Distribution and Adipocyte Biology in Children with Lipodystrophy (LEAD-ABC) study. Participants were stratified into 3 groups: group C, unaffected family members or recruited controls; group A, individuals with a pathogenic variant but without extensive symptoms; and group B, patients with overt FPLD2 (Figure 1A). Whole-genome sequencing identified 5 disease-causing LMNA variants across the 8 pedigrees (Figure 1B). Participants with developing FPLD2 (group A) retained visible subcutaneous WAT on limbs and trunk without visceral WAT accumulation (Figure 1C). In contrast, participants with developed FPLD2 (group B) exhibited classical fat redistribution: loss of subcutaneous WAT, increased visceral adiposity, and prominent fat accumulation in the upper neck and face (Figure 1D). These regional changes were visualized using fat shadow imaging (Figure 1E and Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI198387DS1). Quantitatively, both developing and developed FPLD2 groups had reduced total body fat percentage compared with controls (Figure 1F). Individuals with FPLD2 specifically had reduced leg fat (Figure 1G), and those with developing FPLD2 already had decreased trunk fat (Figure 1H). MRI revealed no significant hepatic fat accumulation in participants with FPLD2 (Figure 1, I and J).
 Figure 1
Figure 1Clinical, metabolic, and molecular characterization of individuals with FPLD2. (A) Study design and patient groups. Groups include unaffected family members (control, group C, n = 6), genetically affected but clinically unaffected individuals (developing, group A, n = 9), and patients with FPLD2 (FPLD2, group B, n = 7). (B) Pedigrees from multiple families with FPLD2. Filled symbols represent affected individuals, open symbols indicate unaffected individuals, and question marks denote unknown phenotypic status. (C) Images of patients with developing FPLD2 with early signs of fat redistribution. (D) Images of patients with FPLD2 phenotypes with peripheral lipoatrophy and upper trunk fat accumulation. (E) Whole-body fat shadows. Quantification of fat mass percentage in (F) total body, (G) leg, and (H) trunk. (I) MRI-based hepatic fat fraction maps with (J) quantification. (K) Hemoglobin A1c (HbA1c) percentages. (L) Triglyceride concentrations in plasma. (M) Nonesterified fatty acid (NEFA) concentrations. (N) Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) scores. (O) Leptin concentrations. (P) Adiponectin concentrations. (Q) Fibroblast growth factor 21 (FGF21) concentrations. Data are represented as mean ± SD. *P < 0.05. Statistical analyses were performed using 1-way ANOVA, followed by Bonferroni’s post hoc test.
We next evaluated metabolic parameters across groups. Participants with developed FPLD2 (group B) exhibited increased glycated hemoglobin (Figure 1K), circulating triglycerides (Figure 1L), nonesterified fatty acids (NEFA) (Figure 1M), and glucose area under the curve (AUC) after oral glucose tolerance test (OGTT) (Supplemental Figure 1B), indicating impaired metabolic control. However, Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) (Figure 1N), insulin AUC (Supplemental Figure 1C), NEFA AUC (Supplemental Figure 1D), fasted insulin (Supplemental Figure 1E), and Adipose Tissue Insulin Resistance Index (ADIPO-IR) (Supplemental Figure 1F) did not differ significantly between groups. Circulating leptin (Figure 1O) and adiponectin (Figure 1P) were reduced in FPLD2, consistent with WAT loss. Fibroblast growth factor 21 (FGF21) (Figure 1Q) and growth differentiation factor 15 (GDF15) (Supplemental Figure 1G) were elevated in FPLD2, consistent with stress or metabolic dysfunction (30). Clinical characteristics are summarized in Table 1. Collectively, these data highlight patterns of adipose loss during FPLD2 progression and confirm that participants with developed, but not developing, FPLD2 exhibit metabolic dysfunction (1, 31).
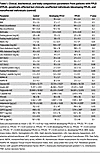 Table 1
Table 1Clinical, biochemical, and body composition parameters from patients with FPLD (FPLD), genetically affected but clinically unaffected individuals (developing FPLD), and nonaffected individuals (control)
WAT biopsies from participants with FPLD2 have decreased fatty acid metabolism and increased inflammation gene expression. To study mechanisms of adipose loss, we collected subcutaneous WAT biopsies from the abdomen (atrophic) and dorsocervical upper neck (expanding) regions (1). Comparing these depots allows within-individual analysis of adipose redistribution in FPLD2. Histological analyses and Picrosirius red staining showed visibly increased fibrosis in abdominal WAT with disease (Figure 2A), though variable sampling limited quantification. Adipocyte size was unchanged in abdominal samples across disease states (Figure 2, B and C). Similar results were observed in the upper neck (Figure 2, D–F). Skin biopsies were similar across groups (Supplemental Figure 1H). These data indicate that lipid-laden adipocytes remain morphologically comparable across disease states.
 Figure 2
Figure 2Biopsies from patients with FPLD2 have no change in adipocyte size, but bulk RNA-sequencing suggests decreased metabolism and increased inflammation in patient adipose tissue. (A) Representative adipose tissue histological images and Picrosirius red–stained tissue for collagens from biopsies across patient groups in abdominal biopsies. Scale bar: 200 μm. (B) Frequency distribution of adipocyte size and (C) frequency of adipocytes less or greater than 2,500 μm2 from abdominal biopsies. (D) Histological images and Picrosirius red analyses on upper neck biopsies. (E) Frequency distribution of adipocyte size and (F) frequency of adipocytes less or greater than 2,500 μm2 from upper neck biopsies. Bulk RNA-sequencing (RNA-Seq) on patient biopsies. Biopsies from group A and B were combined to compare with group C. n = 4–5 samples (upper neck and abdomen combined) per group. Gene set enrichment analysis (GSEA) identified (G) upregulated pathways with normalized enrichment scores (NES) and (H) leading-edge genes for the monocyte chemotaxis pathway. (I) Downregulated GSEA pathways and leading-edge genes for (J) mito protein complex and (K) fatty acid metabolism pathways. Data are represented as mean ± SD. Statistical analyses were performed using 2-way ANOVA, followed by Bonferroni’s post hoc test.
We next examined molecular changes driving FPLD2. Combined abdomen and upper neck biopsies within each group were analyzed by bulk RNA sequencing (RNA-Seq) (Figure 2G). Due to limited sample size, developing and developed FPLD2 groups were combined to analyze broad transcriptomic effects of LMNA variants. Gene Set Enrichment Analysis (GSEA) revealed upregulated inflammation, intermediate filament, and muscle-related pathways (Figure 2, G and H) and downregulated mitochondrial, translational, and fatty acid metabolism pathways (Figure 2, I–K). Fibrosis- and ECM-related genes were altered, including downregulated TMND and upregulated PDGFA, MMP7, MMP16, and multiple collagen genes (Supplemental Figure 1I). Thus, WAT from participants with LMNA variants shows decreased metabolism and increased inflammation, suggesting these contribute to disease progression.
Single nucleus RNA sequencing (snRNA-Seq) identifies depot differences between upper neck and abdominal adipose biopsies. To define depot differences and identify cell types contributing to transcriptomic shifts (Figure 2), we performed snRNA-Seq (10x Genomics) on patient WAT biopsies (n = 4–5), identifying 7 major cell populations: adipocytes, adipose stem and progenitor cells (ASPCs), macrophages, endothelial cells, T cells, pericytes, and lymphatic endothelial cells (LECs) (Figure 3, A and B) (32, 33). Other smaller cell populations such as mast cells, natural killer cells, and dendritic cells were detected but grouped into broader categories for downstream analysis.
 Figure 3
Figure 3Single nucleus RNA-Seq identifies changes in cell proportions and subclustering analyses between subcutaneous abdomen and upper neck adipose tissue in unaffected patients. (A) Uniform manifold approximation and projection (UMAP) of abdomen and upper neck biopsies across all patient groups (A, B, and C). ASPC, adipose stem and progenitor cells; LEC, lymphatic endothelial cells. (B) Marker genes for each cell population. (C) Permutation tests to identify changes in cell proportions in the upper neck relative to the abdominal biopsies from control patients (group C); changes were statistically significant if the log2FC was less or greater than 0.32 and the FDR was less than 0.05. (D) Adipocyte subclusters and corresponding GSEA pathways. (E) Adipocyte subcluster changes between the upper neck and abdomen in control patients. (F) ASPC subclusters with GSEA pathways. EMT, epithelial-mesenchymal transition. (G) ASPC subcluster changes in the upper neck relative to abdomen. (H) Macrophage subclusters with GSEA pathways. (I) Subcluster changes in macrophages in upper neck relative to abdomen. (J) T cell subclusters with GSEA pathways. (K) Subcluster changes in T cells in upper neck relative to abdomen.
In control tissues, the upper neck depot contained more adipocytes and fewer LECs, macrophages, and ASPCs than abdominal WAT (Figure 3C). We next sought to characterize the molecular differences in cell states between depots. Adipocytes formed 2 subclusters: cluster 1 (adipogenic) and cluster 2 (pro-inflammatory) (Figure 3D), with no difference in subcluster proportions between depots (Figure 3E). ASPCs divided into 4 subclusters; the upper neck depot had more adipogenic ASPCs and fewer pro-inflammatory or mTOR-high ASPCs, suggesting higher adipogenic potential in the upper neck depot (Figure 3, F and G).
Among immune cells, lipid-associated macrophages (cluster 3) were decreased in the upper neck versus abdomen (Figure 3, H and I), and low-inflammation T cells (cluster 2) were also reduced (Figure 3, J and K). High-inflammation endothelial cells (cluster 3) were increased in the upper neck (Supplemental Figure 2, A and B), whereas high-myogenesis pericytes and high-translation LECs were decreased (Supplemental Figure 2, C–F). Thus, in healthy individuals, upper neck WAT harbors more pro-adipogenic ASPCs and fewer pro-inflammatory macrophages without altering adipocyte identity.
snRNA-Seq analyses reveal widespread shifts in cellular identity during FPLD2 progression. We next examined how cell type composition changes across disease states. We generated UMAP plots combining both depots across our 3 patient groups (Figure 4A), as well as stratified UMAPs by genotype (Supplemental Figure 3A) and sex (Supplemental Figure 3B). UMAPs combining both depots (Figure 4A) showed reduced adipocyte proportions in abdominal WAT of developing and developed FPLD2 (Figure 4, B and C), consistent with WAT atrophy. LECs increased during developing FPLD2 but declined with progression (Figure 4, B and C). Macrophages and pericytes increased in FPLD2 abdominal WAT (Figure 4C). In the upper neck, adipocytes decreased in developing FPLD2 but stabilized thereafter (Figure 4, D and E). LECs followed a similar transient pattern (Figure 4, D and E). Depot differences seen in controls (Figure 3C) persisted across genotypes (Figure 4, F and G), suggesting intrinsic regional identity.
 Figure 4
Figure 4SnRNA-Seq identifies changes in cell proportions and identity with progression of FPLD2. (A) UMAP of combined upper neck and abdomen biopsies across disease states; cell type markers are the same as in Figure 3. Permutation tests identified changes in cell proportion from abdominal biopsies from patients with (B) developing FPLD2 and (C) FPLD2 relative to control and from upper neck biopsies in (D) developing FPLD2 and (E) FPLD2 relative to control. Cell proportion changes in the upper neck relative to abdomen were identified in (F) developing and (G) FPLD2 disease states. (H) All adipocytes from developing or FPLD2 biopsies were analyzed via GSEA and compared with controls to identify population-level changes in cell identity. Numbers on bar represent the number of genes driving that dataset. Sig, signaling; Org, organization. (I) Adipocyte subcluster analyses with corresponding GSEA pathways (same as in Figure 3) and subcluster changes with disease. (J) ASPC GSEA in developing or FPLD2 biopsies relative to controls. (K) ASPC subcluster changes with disease. (L) Macrophage GSEA in developing or FPLD2 biopsies relative to controls. (M) Macrophage subcluster changes with disease.
After characterizing changes in cell type proportions, we next studied how cell identities change with FPLD2. GSEA of adipocytes showed reduced lipid metabolism and increased ECM and inflammation (Figure 4H), with leading-edge genes listed in Supplemental Table 1. These data align closely with results from bulk RNA-Seq of WAT from participants with FPLD2, which also showed suppression of fatty acid metabolism and increased inflammation (Figure 2, G–K). Adipocyte subcluster composition was unchanged (Figure 4I). ASPCs displayed decreased oxidative phosphorylation and ribosomal genes but increased lipid metabolism (Figure 4J). Pro-adipogenic ASPCs (cluster 2) were expanded, while pro-inflammatory ASPCs (cluster 3) were reduced (Figure 4K). Macrophages overall were less inflammatory (Figure 4L), though lipid-associated macrophages (cluster 3) increased (Figure 4M). Endothelial cell, T cell, pericyte, and LEC populations showed altered metabolic, translational, and inflammatory gene expression (Supplemental Figure 3, C–J). Collectively, adipocytes exhibit impaired fatty acid metabolism, ASPCs adopt a more adipogenic state, and macrophages show lipid-scavenging features during FPLD2 progression.
Cell proportions change with FPLD2 progression in a depot-specific manner. We next assessed how these cell proportion changes differed by depot. In FPLD2, pro-inflammatory adipocytes were enriched in the upper neck relative to the abdomen (Supplemental Figure 4A). Pro-adipogenic ASPCs increased in developing FPLD2 (Supplemental Figure 4B), possibly explaining dorsocervical WAT expansion. Lipid-associated macrophages were reduced in the upper neck (Supplemental Figure 4C), pro-inflammatory endothelial cells were increased in the upper neck (Supplemental Figure 4D), and pro-inflammatory T cells rose in developing FPLD2 (Supplemental Figure 4E). Pericytes were unchanged (Supplemental Figure 4F), and pro-adipogenic LECs were increased in early disease (Supplemental Figure 4G). Thus, depot-specific cellular shifts, particularly increased pro-inflammatory adipocytes and endothelial cells in the upper neck, may underlie the contrasting fat redistribution and metabolic features of FPLD2.
Inducible lamin A/C knockout in adipocytes causes lipodystrophy but not metabolic dysfunction. Bulk RNA-Seq and snRNA-Seq data from participants with FPLD2 revealed downregulation of metabolic pathways and upregulation of inflammatory signaling (Figures 2 and 4). To further investigate roles of lamin A/C in adipocyte maintenance, we generated LmnaiADKO mice, extending our prior constitutive adiponectin-Cre (Adipoq-Cre) model (28). Tamoxifen was administered intraperitoneally for 5 days to Lmnafl/fl and LmnaiADKO mice (Figure 5A). Two weeks posttamoxifen, LmnaiADKO mice exhibited reduced fat mass (Figure 5B) without changes in body weight or lean mass (Supplemental Figure 5, A and B). Posterior subcutaneous (psWAT) and epididymal WAT (eWAT) weights decreased at 2 to 4 weeks posttamoxifen, partially recovering by 16 weeks (Figure 5, C and D). Retroperitoneal WAT decreased at 2 weeks posttamoxifen; brown adipose tissue (BAT), perirenal WAT, and liver were unchanged (Supplemental Figure 5C). No sex differences were observed (Supplemental Figure 5D), and both sexes were used throughout these mouse studies. Histology revealed no overt WAT or liver changes (Figure 5E and Supplemental Figure 5F), though slight BAT whitening and partial bone marrow adipose loss appeared by 8 weeks posttamoxifen. Uncoupling protein 1 expression was unchanged in LmnaiADKO mouse BAT 2 weeks posttamoxifen and undetectable in psWAT (Supplemental Figure 5, F and G). Circulating adiponectin decreased at 4 weeks posttamoxifen (Figure 5F and Supplemental Figure 5H), but insulin sensitivity and glucose tolerance remained normal at 6 and 12 weeks posttamoxifen (Figure 5, G and H, and Supplemental Figure 5, I and J). LmnaiADKO mice thus model early adipocyte loss without confounding metabolic dysfunction, ideal for mechanistic studies.
 Figure 5
Figure 5Tamoxifen-inducible adipocyte-specific Lmna knockout causes transient adipose tissue loss, and Lmna-deficient adipocytes shrink and disappear. All data from male mice besides histology from female mice. (A) Gene schematic of Lmnafl/fl control mice and LmnaiADKO mice. Adult mice were administered tamoxifen intraperitoneally for 5 consecutive days to induce recombination. (B) Fat mass after tamoxifen administration (n = 6). (C) Posterior subcutaneous WAT (psWAT) weights and (D) epididymal WAT (eWAT) weights at 2, 4, and 16 weeks posttamoxifen (n = 3–6). (E) Representative histology of psWAT and parametrial WAT (pmWAT) 2 weeks posttamoxifen. Scale bar: 40 μm. (F) Serum adiponectin immunoblot at 0 and 4 weeks posttamoxifen (n = 4–5). (G) Insulin tolerance test 6 weeks posttamoxifen (n = 6). (H) Glucose tolerance test 7 weeks posttamoxifen (n = 6). (I) Schematic of mTmG reporter system induced by tamoxifen-mediated Cre activity. (J) Representative fresh confocal micrographs of psWAT (scale bar: 100 μm) and (K) quantification of psWAT adipocyte size at 2 weeks posttamoxifen (n = 3–4). (L) Quantification of psWAT GFP+ or tdTomato+ adipocytes. (M) Confocal micrographs of eWAT and (N) quantification of eWAT adipocyte size at 2 weeks posttamoxifen. (O) Quantification of eWAT GFP+ or tdTomato+ adipocytes. Data are represented as mean ± SD. *P < 0.05. Statistical analyses were performed using 2-way ANOVA, followed by Bonferroni’s post hoc test.
Lamin A/C-deficient adipocytes shrink, become misshapen, and disappear from WAT. Using the mTmG reporter system (34), we tracked Lmna-KO adipocytes via GFP expression (Figure 5I). Two weeks posttamoxifen, GFP+ adipocytes in psWAT were widespread and morphologically normal despite reduced fat mass, suggesting fewer adipocytes (Figure 5, J–L). By 6 weeks posttamoxifen, Lmna-KO GFP+ adipocytes showed shrinkage, irregular shape, and membrane budding, while tdTomato+ cells increased, suggesting compensatory adipogenesis (Figure 5, J and L). By 16 weeks posttamoxifen, GFP+ adipocytes were nearly absent in psWAT from LmnaiADKO mice. This same pattern was observed in eWAT (Figure 5, M–O). Small GFP+ cells in WAT at 6 weeks posttamoxifen disappeared by 16 weeks, suggesting that KO adipocytes do not persist in LmnaiADKO WAT (Supplemental Figure 6, A and B). Flow cytometry confirmed GFP+ stromal vascular cells (SVCs) were not elevated at 6 weeks posttamoxifen in LmnaiADKO WAT, indicating no evidence of dedifferentiation of KO adipocytes (Supplemental Figure 6, D and E). These data indicate Lmna-KO adipocytes progressively atrophy and are cleared from tissue, supporting lamin A/C’s essential role in adipocyte maintenance.
LmnaiADKO WAT mirrors FPLD2 WAT: increased inflammation, decreased fatty acid metabolism. We performed bulk RNA-Seq and proteomics on pmWAT 2 weeks posttamoxifen, prior to morphological changes, and integrated GSEAs between both datasets (Figure 6A). GSEA revealed upregulated immune processes (myeloid activation, antigen binding) and downregulated oxidative phosphorylation and fatty acid biosynthesis (Figure 6B). Proteomics specifically showed elevated cell death and suppressed muscle-associated pathways (Figure 6C). Lipogenic genes and mitochondrial genes were repressed (Figure 6, D and E), while inflammation and cell death genes increased (Figure 6, F and G). Comparison with constitutive LmnaADKO WAT showed concordant suppression of metabolism and increased inflammation (Supplemental Figure 7, A–D). Integration with human FPLD2 RNA-Seq verified overlapping gene expression patterns: increased inflammation and decreased mitochondrial/lipid metabolism pathways (Figure 6, H–J), highlighting lamin A/C’s role in adipocyte homeostasis.
 Figure 6
Figure 6Bulk RNA-Seq and proteomics of LmnaiADKO WAT reveals increased inflammation and reduced fatty acid metabolism and mitochondrial pathways, similar to human FPLD2 biopsies. All data from female mice. LmnaiADKO and Lmnafl/fl pmWAT 2 weeks posttamoxifen was used for RNA-Seq (n = 6–7) and proteomics (n = 5). (A) Integrative GSEA on bulk RNA-Seq and proteomics. Pink dots symbolize that NES significantly changed in both RNA-Seq and proteomics datasets, orange dots that NES changed only in proteomics, and blue that NES changed only in RNA-Seq. (B) Highlighted GSEA pathways changed in both proteomics and RNA-Seq. Numbers on bars represent number of overlapping genes driving pathways. (C) Highlighted GSEA pathways changed in proteomics only. Heatmaps showing selected changes in genes driving (D) lipid biosynthesis, (E) mitochondrial function, (F) inflammation, and (G) cell death. TAG, triacylglycerol. (H) Integrative GSEA of LmnaiADKO pmWAT bulk RNA-Seq compared with FPLD2 bulk RNA-Seq (Figure 2). (I) Highlighted GSEA pathways changed in both mouse and human RNA-Seq datasets. (J) Leading-edge genes for GSEA pathways related to inflammation, fatty acid metabolism, and mitochondrial function for either iADKO or FPLD2 samples.
Lipogenic and mitochondrial protein expression is lower in LmnaiADKO WAT, accompanied by decreased respiration and altered mitochondrial structure. In LmnaiADKO psWAT, which showed no change in mass 2 weeks posttamoxifen (Figure 5C), PPARγ and C/EBPα were unchanged in protein expression, whereas ChREBP and key lipogenic enzymes (ACC, FASN, SCD1) were reduced (Figure 7A and Supplemental Figure 8A). eWAT showed similar reductions, with slightly increased PPARγ, possibly compensatory (Figure 7B and Supplemental Figure 8B). Given the well-established link between lipid metabolism and mitochondrial function (35, 36), we examined mitochondrial protein expression and saw that oxidative phosphorylation proteins were reduced in LmnaiADKO eWAT (Figure 7C and Supplemental Figure 8C), with decreased baseline, maximal, and ATP-linked respiration; spare respiratory capacity; and proton leak in pmWAT adipocytes (Figure 7, D and E, and Supplemental Figure 8, D–F). In contrast, Lmna-KO adipocytes isolated from psWAT at the same time point did not exhibit changes in mitochondrial respiration (Supplemental Figure 8, G–K), suggesting that reductions in lipid metabolism proteins (Figure 7A) may precede overt mitochondrial dysfunction following Lmna deletion in adipocytes. Mitochondrial DNA content remained unchanged in psWAT and pmWAT (Supplemental Figure 8, L and M). Mitochondrial biogenesis and fission-fusion regulators (PGC1α, MFN2, OPA1, VDAC1) were decreased in expression in LmnaiADKO adipocytes; TOMM20 was unchanged (Figure 7F and Supplemental Figure 8, N and O). Imaging revealed irregular mitochondrial clustering and polarization in Lmna-KO adipocytes compared with controls (37), a potential sign of cell damage, as previously observed in MFN2-KO cells (38) (Figure 7G). Transmission electron microscopy (TEM) of LmnaiADKO WAT 2 weeks posttamoxifen showed small lipid droplets with surrounding mitochondria, suggesting active lipid synthesis, droplet fission, or budding (Figure 7H). Cristae were disorganized and adipocyte had potentially altered heterochromatin distribution, though overall mitochondrial area and droplet contacts were unchanged (Figure 7, H–N). Deletion of Lmna in adipocytes thus impairs mitochondrial function and structure, contributing to adipocyte loss.
 Figure 7
Figure 7LmnaiADKO adipocytes have reduced oxygen consumption and dysfunctional mitochondrial dynamics and structure. Data in A to C and in H are from male mice, and all other data are from female mice. Immunoblot analyses 2 weeks posttamoxifen of lipid metabolism and lipogenesis proteins in (A) psWAT and (B) eWAT (n = 4–5). Loading control = laminin. (C) Immunoblot of mitochondrial proteins in eWAT 2 weeks posttamoxifen (n = 4–5). Loading control = laminin. Oroboros Oxygraph-2k analyses of (D) baseline and (E) maximum respiration in floated adipocytes from pmWAT 2 weeks posttamoxifen (n = 6). (F) Immunoblot of mitochondrial function and dynamics proteins in floated adipocytes and stromal vascular fraction (SVF) from pmWAT 2 weeks posttamoxifen (n = 3–5). * indicates nonspecific band. Loading control = laminin and β-actin. (G) Confocal micrographs of floated adipocytes from pmWAT stained for nuclei (Hoechst), lipid (BODIPY), and mitochondria (MitoTracker Red); scale bar: 50 μm. (H) Transmission electron micrographs of eWAT 2 weeks posttamoxifen. Scale bar: 1 μm for original magnification, 3,000×, images; 200 nm for original magnification, 10,000×, images. N, nucleus; L, lipid droplet; M, mitochondria. Quantification of transmission electron micrographs: (I) distance of mitochondria to closest lipid droplet, (J) mitochondrial area, (K) cristae volume per mitochondria, (L) mitochondrial circularity, and (M) cristae anisotropy with (N) example of anisotropy analysis. An average of 9 adipocyte mitochondria were quantified per mouse; n = 5–6. Data are represented as mean ± SD. *P < 0.05. Statistical analyses were performed using Student’s t test.
Adipocyte loss is not driven by increased lipolysis. Following our TEM observations of increased small lipid droplets in LmnaiADKO adipocytes, we investigated whether enhanced lipolysis might cause adipocyte loss in vivo. Circulating glycerol decreased at 4 weeks posttamoxifen and later, under fed and fasted conditions in LmnaiADKO mice (Supplemental Figure 9A). Isoproterenol-stimulated lipolysis was unchanged when normalized to fat mass at 4 weeks posttamoxifen in LmnaiADKO mice (Supplemental Figure 9, B and C). Bulk RNA-Seq showed downregulation of lipolytic genes at 2 weeks posttamoxifen (Supplemental Figure 9D), but lipolytic proteins were not suppressed (Supplemental Figure 9, E and F), indicating adipocyte loss occurs via lipolysis-independent mechanisms.
Lmna-deficient adipocytes show cell-autonomous pro-inflammatory gene expression. snRNA-Seq in human FPLD2 suggested immune signatures arise partly from macrophages and T cells. Spectral flow cytometry of LmnaiADKO psWAT and pmWAT 2 weeks posttamoxifen showed no differences in total SVCs, CD45+ cells, adipose tissue macrophages (ATMs; CD64+), or identity of macrophages (CD11c+, TIM4+, CD163+) or T cells (CD4+, CD8+) (Supplemental Figure 10, A–N). Whole WAT from LmnaiADKO mice had few inflammatory transcript changes (Supplemental Figure 10, O and P), but isolated Lmna-KO adipocytes 2 weeks posttamoxifen showed elevated Il6, Il10, Nlrp3, Il1b, and Tnfa, whereas SVF had minor changes (Supplemental Figure 10, Q–T), suggesting cell-autonomous inflammatory signaling upon loss of lamin A/C. Cleaved caspase-3 was undetectable at 2, 4, and 6 weeks posttamoxifen in LmnaiADKO WAT, and cGAS-STING markers were unchanged (Supplemental Figure 10, U–W), indicating a nonclassical, asynchronous cell death mechanism.
Fundamental role of lamin A/C in regulating lipid metabolism and inflammatory gene expression across cell types. We observed consistent gene expression changes in Lmna-deficient mouse adipocytes and human FPLD2 WAT: suppressed lipid metabolism and increased inflammation. To assess whether these changes are conserved across cell types, we analyzed publicly available microarray and assay for transposase-accessible chromatin sequencing (ATAC-Seq) data from Lmna-KO MEFs, which also showed impaired mitochondrial respiration and irregular mitochondrial localization (Figure 8A) (39, 40). Integration of LmnaiADKO WAT RNA-Seq with Lmna-KO MEF datasets revealed coordinated dysregulation of lipid metabolism and inflammation at transcript and chromatin levels (Figure 8, A–E). Overlapping genes included Acsl4, Acsl6, Rbp1, and Scl27a3 (lipid homeostasis) and Il1rl1, Il33, and Cd44 (inflammation), suggesting lamin A/C directly regulates these programs across cell types.
 Figure 8
Figure 8Lmna-KO MEFs and adipocytes have shared patterns of altered gene expression, correlated with changes in chromatin accessibility. (A) Schematic of dataset integration between LmnaiADKO pmWAT RNA-Seq and Lmna-KO MEF microarray (39, 40) and assay for transposase-accessible chromatin sequencing (ATAC-Seq). Overlapping changed genes in RNA-Seq of LmnaiADKO WAT (iADKO), Lmna-KO MEF microarray (MEF), and Lmna-KO MEF ATAC-Seq (ATAC) were identified, and GSEA was run to identify changes in (B) lipid metabolism Gene Ontology (GO) terms with their corresponding (C) overlapping leading-edge genes, along with (D) inflammatory GO terms with (E) corresponding overlapping leading-edge genes. (F) Distribution of chromatin features represented by percentage of overall chromatin composition compared between Lmna control (CTRL) and KO MEF ATAC-Seq. (G) Enriched motifs in Lmna-KO MEFs, stratified into 4 bins based on microarray gene expression changes, from most downregulated to most upregulated. (H and I) ATAC-Seq signal tracks and visualization of significantly enriched regions and their predicted target genes. Red lines represent enhancers with a positive fold-change upon Lmna KO, and blue lines represent negative.
ATAC-Seq peak distribution across chromatin features was unchanged between control and KO MEFs (Figure 8F), consistent with lamin A/C binding ~30%–40% of the genome (41, 42). Motif enrichment analysis near differentially expressed genes revealed KLF family motifs enriched in downregulated genes and HNF1A/B, HOX, and FOS/JUN motifs near upregulated genes (Figure 8G). Klf4 and Klf13 loci showed altered enhancer accessibility and reduced expression (Figure 8, H and I). KLF4 regulates adipocyte differentiation (44), and KLF13 has been implicated in suppressing inflammation (45), suggesting loss of lamin A/C impairs adipocyte function and enhances inflammatory signaling. Together, these data indicate that lamin A/C loss disrupts enhancer accessibility and transcriptional regulation of lipid- and inflammation-related genes across diverse cell types, highlighting a conserved role in maintaining metabolic and immune gene programs, beyond adipocytes or FPLD2.












Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)