Advertisement
Review Series
Open Access | ![]() 10.1172/JCI191944
10.1172/JCI191944
Evolving concepts in adjuvant/neoadjuvant therapy for resectable pancreas cancer
John M. Bryant,1 Luis Ruffolo,2 Kevin Soares,2 Sarah Hoffe,1 and Andrew M. Lowy3
1Department of Radiation Oncology, Section of Gastrointestinal Radiation Oncology, Moffitt Cancer Center, Tampa, Florida, USA.
2Department of Surgery, Hepatopancreatobiliary Service, Memorial Sloan Kettering Cancer Center, New York, New York, USA.
3Department of Surgery, Division of Surgical Oncology, Moores Cancer Center, University of California San Diego, La Jolla, California, USA.
Address correspondence to: Sarah Hoffe, 12902 Magnolia Drive, Tampa, Florida 33612, USA. Email: sarah.hoffe@moffitt.org. Or to: Kevin Soares, 1275 York Ave, C887, New York, New York 10065, USA. Email: soaresk@mskcc.org. Or to: Andrew M. Lowy, 3855 Health Sciences Drive #0987, La Jolla, California 92093, USA. Email: alowy@health.ucsd.edu.
Authorship note: JMB and LR contributed equally to this work.
Find articles by Bryant, J. in: PubMed | Google Scholar
1Department of Radiation Oncology, Section of Gastrointestinal Radiation Oncology, Moffitt Cancer Center, Tampa, Florida, USA.
2Department of Surgery, Hepatopancreatobiliary Service, Memorial Sloan Kettering Cancer Center, New York, New York, USA.
3Department of Surgery, Division of Surgical Oncology, Moores Cancer Center, University of California San Diego, La Jolla, California, USA.
Address correspondence to: Sarah Hoffe, 12902 Magnolia Drive, Tampa, Florida 33612, USA. Email: sarah.hoffe@moffitt.org. Or to: Kevin Soares, 1275 York Ave, C887, New York, New York 10065, USA. Email: soaresk@mskcc.org. Or to: Andrew M. Lowy, 3855 Health Sciences Drive #0987, La Jolla, California 92093, USA. Email: alowy@health.ucsd.edu.
Authorship note: JMB and LR contributed equally to this work.
Find articles by Ruffolo, L. in: PubMed | Google Scholar
1Department of Radiation Oncology, Section of Gastrointestinal Radiation Oncology, Moffitt Cancer Center, Tampa, Florida, USA.
2Department of Surgery, Hepatopancreatobiliary Service, Memorial Sloan Kettering Cancer Center, New York, New York, USA.
3Department of Surgery, Division of Surgical Oncology, Moores Cancer Center, University of California San Diego, La Jolla, California, USA.
Address correspondence to: Sarah Hoffe, 12902 Magnolia Drive, Tampa, Florida 33612, USA. Email: sarah.hoffe@moffitt.org. Or to: Kevin Soares, 1275 York Ave, C887, New York, New York 10065, USA. Email: soaresk@mskcc.org. Or to: Andrew M. Lowy, 3855 Health Sciences Drive #0987, La Jolla, California 92093, USA. Email: alowy@health.ucsd.edu.
Authorship note: JMB and LR contributed equally to this work.
Find articles by Soares, K. in: PubMed | Google Scholar
1Department of Radiation Oncology, Section of Gastrointestinal Radiation Oncology, Moffitt Cancer Center, Tampa, Florida, USA.
2Department of Surgery, Hepatopancreatobiliary Service, Memorial Sloan Kettering Cancer Center, New York, New York, USA.
3Department of Surgery, Division of Surgical Oncology, Moores Cancer Center, University of California San Diego, La Jolla, California, USA.
Address correspondence to: Sarah Hoffe, 12902 Magnolia Drive, Tampa, Florida 33612, USA. Email: sarah.hoffe@moffitt.org. Or to: Kevin Soares, 1275 York Ave, C887, New York, New York 10065, USA. Email: soaresk@mskcc.org. Or to: Andrew M. Lowy, 3855 Health Sciences Drive #0987, La Jolla, California 92093, USA. Email: alowy@health.ucsd.edu.
Authorship note: JMB and LR contributed equally to this work.
Find articles by Hoffe, S. in: PubMed | Google Scholar
1Department of Radiation Oncology, Section of Gastrointestinal Radiation Oncology, Moffitt Cancer Center, Tampa, Florida, USA.
2Department of Surgery, Hepatopancreatobiliary Service, Memorial Sloan Kettering Cancer Center, New York, New York, USA.
3Department of Surgery, Division of Surgical Oncology, Moores Cancer Center, University of California San Diego, La Jolla, California, USA.
Address correspondence to: Sarah Hoffe, 12902 Magnolia Drive, Tampa, Florida 33612, USA. Email: sarah.hoffe@moffitt.org. Or to: Kevin Soares, 1275 York Ave, C887, New York, New York 10065, USA. Email: soaresk@mskcc.org. Or to: Andrew M. Lowy, 3855 Health Sciences Drive #0987, La Jolla, California 92093, USA. Email: alowy@health.ucsd.edu.
Authorship note: JMB and LR contributed equally to this work.
Find articles by Lowy, A. in: PubMed | Google Scholar
Published July 15, 2025 - More info
J Clin Invest. 2025;135(14):e191944. https://doi.org/10.1172/JCI191944.
© 2025 Bryant et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
-
Neoadjuvant versus adjuvant therapy for resectable PDAC
Given the high rate of distant relapse after surgical resection of PDAC, systemic therapy is essential for curative treatment. Phase III studies have shown the benefit of adjuvant chemotherapy in prolonging disease-free survival (DFS) and overall survival (OS) over surgery alone, with regimens including FOLFIRINOX (folinic acid, fluorouracil, irinotecan, and oxaliplatin), gemcitabine/capecitabine, and gemcitabine/nab-paclitaxel (G-NP) improving OS over gemcitabine alone (1–5). An important consideration is that patients enrolled in adjuvant therapy trials represent a highly selected group, having successfully recovered from surgery. Neoadjuvant chemotherapy has been driven by several ideas: (a) Treating occult metastatic disease early may improve outcomes. (b) Pancreatic surgery is morbid, and postoperative complications may delay or prevent adjuvant therapy. Delivering therapy before surgery ensures chemotherapy exposure. (c) Chemotherapy prior to surgery is better tolerated. (d) Neoadjuvant therapy allows for response assessment. (e) Patients with refractory disease may avoid non-therapeutic surgery. (f) Preoperative therapy provides observation time to manage comorbidities and optimize patient fitness for successful surgery recovery.
Remarkably, even though neoadjuvant therapy has been delivered for decades, there remains extremely limited level 1 data directly comparing neoadjuvant to adjuvant therapy. Most studies of “neoadjuvant therapy” have used perioperative chemotherapy rather than total neoadjuvant chemotherapy. Table 1 summarizes landmark studies utilizing perioperative chemotherapy for PDAC (6). Several conclusions can be drawn. Patients treated with perioperative chemotherapy on average receive a greater percentage of intended chemotherapy in the preoperative period rather than the adjuvant setting. Most studies incorporating neoadjuvant therapy also demonstrated a reduction in positive lymph nodes and found an improvement in rates of margin-negative (R0) resection, both prognostic factors for OS.
Neoadjuvant therapy has many hypothesized advantages, but it poses multiple clinical challenges. These include the need for preoperative tissue diagnosis, preoperative biliary drainage in jaundiced patients, and venous access for chemotherapy. Monitoring patients during chemotherapy requires a coordinated multidisciplinary team to swiftly address treatment-related toxicity and biliary stent complications. Failure to address these issues may threaten patients’ candidacy for surgical resection. The NORPACT-1 phase II trial reported superior survival in patients undergoing pancreatectomy and adjuvant chemotherapy compared with intended 4 cycles of neoadjuvant FOLFIRINOX and adjuvant therapy (10). The study has faced numerous critiques, further emphasizing the need for level 1 data in this area. Two identically designed phase III trials comparing perioperative FOLFIRINOX (four cycles of neoadjuvant and two cycles of adjuvant therapy) are nearing full accrual in the United States (Alliance A021806) and Europe (PREOPANC-4) (Table 1). These studies will offer new insights into the value of these approaches for managing patients with resectable PDAC. Despite the results, given PDAC’s known inter- and intrapatient heterogeneity, many questions will remain — questions tracing back to our limited understanding of how systemic therapies impact PDAC biology. Major unanswered questions include:
Can we identify biomarkers to select patients who should receive neoadjuvant therapy versus undergoing upfront surgical resection? Often clinicians will select neoadjuvant therapy in those they judge to be at high risk for rapid relapse; however, specification of the prognostic factors predictive of rapid relapse remains imprecise. Molecular diagnostics such as circulating tumor DNA (ctDNA) are under active investigation to more precisely define radiographically occult disease. Early studies clearly suggest that detection of ctDNA prior to surgery and after neoadjuvant therapy are negative prognostic factors for resection. For example, data from the PANACHE01-PRODIGE48 trial demonstrated a median OS of 19.4 months in patients with a preoperative carbohydrate antigen 19-9 (CA19-9) level greater than 80 U/mL who also had detectable ctDNA and 30.2 months in the CA19-9–high or ctDNA+ group, and OS was not reached in the CA19-9–low or ctDNA– group (log-rank P = 0.0069) (11). Similarly, in a study of resected PDAC patients, the median relapse-free survival was 13 months for patients in whom postoperative ctDNA was positive versus 22 months for those with negative ctDNA (P = 0.003) (12). Such data suggest that we may be able to define parameters predictive of very poor outcomes from surgery; such patients would be ideal candidates for neoadjuvant therapy using novel therapeutic approaches, among them vaccine strategies discussed in the next section. However, low sensitivity and thus a poor negative predictive value severely limit the utility of ctDNA at present (13).
What is the optimal duration of neoadjuvant/adjuvant therapy? Typically, preoperative therapy duration is guided by patient tolerance and clinical measures of response, including radiographic response and CA19-9 when detectable. No prospective studies have addressed this question specifically, while retrospective studies suggest that longer durations of neoadjuvant therapy are associated with better outcomes. However, these findings are biased, as treatment-related toxicity or ineffectiveness usually causes therapy to stop. Our inability to accurately define clinical benefit from neoadjuvant therapy remains a large gap that is currently being addressed by molecular diagnostics and imaging strategies. The most straightforward approach uses PET scans to assess for clinical response. A Mayo Clinic group reported that PET responses were prognostic for improved outcomes after neoadjuvant therapy (14). Other approaches under investigation include radiomics and molecular diagnostics like ctDNA (15). Improving our ability to understand the clinical benefit of neoadjuvant therapy in real time will help us individualize treatment duration more precisely, improving survival and reducing treatment-related toxicity.
Recently, SWOG 1505 noted greater median dose density of both modified FOLFIRINOX (mFOLFIRINOX) and G-NP when received preoperatively versus postoperatively (mFOLFIRINOX preoperative 87.5% vs. postoperative 59.6%, P < 0.001; G-NP preoperative 77.3% vs. postoperative 51.7%, P < 0.001) (7). In this study, dose density received was associated with median survival (8). Similarly, a recent retrospective single-institution study of 225 patients who underwent pancreatectomy for stage I/II PDAC found that regardless of treatment sequence, completion of at least 67% of the recommended number of chemotherapy cycles was associated with improved OS (median OS, 34.5) compared with less than 67% of cycles (median OS, 17.9 months; HR, 0.39; 95% CI, 0.24–0.64) (9). However, neoadjuvant therapy was associated with a greater likelihood of receiving more than 67% of prescribed cycles of chemotherapy. An analysis of ESPAC-3 data demonstrated that completion of all six cycles of planned adjuvant chemotherapy rather than early initiation was an independent prognostic factor for survival after resection (16). A recent retrospective study examined the duration of adjuvant chemotherapy after preoperative FOLFIRINOX and found that adjuvant treatment improved survival (17). The effect was most pronounced in patients receiving less than 4 months of preoperative therapy, consistent with prior data suggesting that receipt of a minimum of two-thirds of prescribed therapy is beneficial.
Is it beneficial to switch neoadjuvant chemotherapy? For patients who receive perioperative therapy, a common clinical dilemma involves determining criteria for changing therapy either during the neoadjuvant component or in the adjuvant setting. There are prospective data to guide such decisions, but a recent meta-analysis of five retrospective studies involving 863 patients who underwent neoadjuvant therapy for localized PDAC found that 20% of patients underwent chemotherapy switching (18). Of these, 42% underwent curative-intent resection, and their survival was comparable to that of patients receiving first-line chemotherapy. Three phase II trials (NCT03322995, NCT04594772, and NCT04539808, ClinicalTrials.gov) are currently in progress in the United States evaluating chemotherapy switching for patients with potentially resectable PDAC. All are non-randomized phase II studies using FOLFIRINOX, each with slightly different criteria for switching to G-NP. While these studies will provide additional new data, ultimately only a randomized trial can definitively address the question of whether changing therapy in the neoadjuvant setting is of benefit, and unfortunately these studies’ relevance may be overrun by the emergence and integration of predictive biomarkers and more effective systemic therapies, namely KRAS inhibitors, as discussed in the next section.
How will our evolving understanding of PDAC biology and KRAS inhibitors change our approach to adjuvant/neoadjuvant therapy? Identifying basal and classical transcriptional subtypes of PDAC has led to studies on whether this biology predicts response or resistance to systemic therapies (19). Early data suggested that the basal subtype may be more resistant to FOLFIRINOX versus G-NP, with ongoing studies of biomarker-selected neoadjuvant therapy, including one using the Purity Independent Subtyping of Tumors (PurIST) classifier to differentiate basal versus classical subtypes (NCT0468331) (20). Recent preclinical data suggest that PDAC has distinct tumor-intrinsic kinomes related to basal and classical subtyping, which have implications for therapeutic response (21). For instance, basal-subtype tumors were more reliant on EGFR signaling and thus more responsive to EGFR inhibitors. Emerging data suggest that classical-subtype tumors may be more resistant to KRAS inhibition, a hypothesis that needs clinical testing (22). With pan-KRAS and KRASG12D-specific inhibitors entering late-phase trials for advanced disease, the next frontier will be their incorporation into the treatment of resectable disease. Given that response rates in chemorefractory advanced PDAC have ranged from 20% to 30% and disease control rates have approached 90%, it is hypothesized that KRAS inhibitors will markedly improve outcomes for resectable disease (23, 24). We need to understand whether there is a biological rationale for timing these therapies relative to surgical resection. Effective therapy prior to an operation could improve margin-negative surgery and reduce procedure-related tumor cell dissemination. Alternatively, reducing tumor cell burden via surgery may enhance the effectiveness of adjuvant KRAS inhibitors. Addressing such questions with preclinical studies is challenging, necessitating next-generation clinical trials that integrate these biomarkers to understand the place of these emerging therapies in managing patients with resectable PDAC, and that integrate them with cytotoxic regimens with proven, though modest, benefit.

-
Radiation therapy for borderline or locally advanced PDAC
For patients with borderline or locally advanced unresectable PDAC, achieving resection is crucial for survival. Only 15%–20% of locally advanced pancreatic cancer (LAPC) patients and 50%–60% of borderline resectable pancreatic cancer (BRPC) patients undergo resection, but they achieve outcomes comparable to those of initially resectable patients if R0 resection is achieved (34–42). The PREOPANC-1 trial provided randomized evidence for neoadjuvant radiotherapy in resectable and borderline resectable PDAC. This phase III trial randomized 246 patients to receive either gemcitabine-based CRT (36 Gy in 15 fractions) followed by surgery and adjuvant gemcitabine, or upfront surgery with adjuvant gemcitabine. Results showed a slight improvement in median survival (15.7 vs. 14.3 months, P = 0.025) and a higher rate of R0 resections (72% vs. 43%, P < 0.001) (43). However, subgroup analysis showed no survival benefit for resectable PDAC, and the use of an outdated chemotherapy regimen limits broader applicability. As a result, many centers consider both upfront surgery followed by chemotherapy and neoadjuvant radiotherapy viable for resectable PDAC (44).
Neoadjuvant radiotherapy is typically delivered using conventional fractionation over 5 to 6 weeks (45–51). Standard fractionation with BED10 less than 70 Gy has not shown significant survival benefits for unresectable PDAC, prompting investigation of hypofractionated radiotherapy (HFRT) and stereotactic body radiotherapy (SBRT) as alternatives to deliver higher BED10 regimens up to 100 Gy (52–56). The interval from standard fractionation radiotherapy to surgery allows more time for disease progression. A 2-week accelerated fractionation schedule, incorporating an intraoperative boost, was explored to shorten treatment time (57, 58). This accelerated method reduced overall treatment duration and applied HFRT in PDAC early.
Modern image-guided radiotherapy techniques have allowed safe dose escalation while reducing treatment sessions. HFRT relies on daily imaging to ensure proper setup. SBRT, delivered in five or fewer fractions, is well suited for PDAC regimens. Shorter treatment durations help reduce delays to surgery or systemic therapy, and stereotactic techniques enable safer delivery of higher doses. Despite improvements, the role of these modern approaches in PDAC treatment remains debated, highlighting the need for innovation and well-designed clinical trials (59–63).
Stereotactic magnetic resonance–guided adaptive radiation therapy (SMART) integrates MRI guidance with adaptive planning, offering real-time tumor tracking and daily plan adjustments. SMART delivers ablative doses while minimizing toxicity to surrounding structures (64). Clinical data on SMART for BRPC and LAPC are encouraging. In a study by Rudra et al., ablative SMART improved local control and OS compared with non-adaptive SBRT in unresectable disease (65). A single-center study reported a 96% R0 resection rate following SMART, with no grade 2+ acute toxicities and excellent postoperative outcomes (66). Median progression-free survival exceeded 13 months, underscoring SMART’s potential to enhance surgical and oncologic outcomes.
Two recent studies exploring SMART in upfront unresectable PDAC after induction chemotherapy showed promising efficacy and safety. A Danish phase II trial with 28 LAPC patients reported a 21% resection rate and a median OS of 20.8 months, which was improved by 7.7 months in those resected (37). The phase II multicenter SMART trial (NCT03621644) enrolled 136 upfront unresectable PDAC patients, showing a 22.8-month median OS and a 94% one-year OS rate (67). SMART was well tolerated in both trials, aligning with retrospective reports (65, 69–71).
Ablative dose radiation continues to garner support for continued exploration in prospective randomized trials after neoadjuvant chemotherapy for upfront unresectable PDAC.
Active clinical trials exploring novel approaches incorporating radiation therapy and surgery in PDAC are highlighted in Table 2. The PANDAS–PRODIGE 44 trial (NCT02676349) evaluates BRPC patients randomized to receive either neoadjuvant modified FOLFIRINOX alone or with conventional CRT at 50.4 Gy with capecitabine.
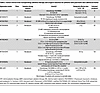 Table 2
Table 2Active clinical trials incorporating radiation therapy and surgical resection for patients with pancreatic duct adenocarcinoma
Tailoring patient selection for radiotherapy may enhance outcomes while minimizing toxicity. Mutations in DNA damage response (DDR) pathways are linked to improved outcomes with platinum-based chemotherapies and PARP inhibitors in PDAC, though their impact on radiotherapy response is less well understood (72). Early data suggest increased radiosensitivity in DDR tumors in preclinical models and retrospective studies (73, 74). Future studies targeting patients with DDR mutations will be essential to evaluate whether incorporating radiotherapy into multimodal treatments can enhance outcomes.
The transcription factor NRF2, often upregulated in PDAC as a result of KRAS mutations, contributes to chemotherapy and radiotherapy resistance by activating antioxidant DNA response elements and reducing reactive oxygen species, critical mediators of radiation-induced DNA damage (75). Upregulated NRF2 expression is associated with poorer survival rates in patients receiving radiation therapy (76). Strategies inhibiting NRF2 or its pathways, like glutamine metabolism, have shown potential in preclinical models to enhance sensitivity to radiation and chemotherapy (75, 76). These findings highlight opportunities to address resistance mechanisms in PDAC, expanding the pool of resectable patients after neoadjuvant therapy and improving overall outcomes in this challenging disease.
In summary, trials incorporating non-ablative doses of radiotherapy have failed to show significant impact and have caused controversy regarding the benefit of radiotherapy in the treatment of PDAC. Recent innovations in the precision of radiation oncology delivery techniques have led to safe dose escalation, suggesting that local-control improvements will only be identified if the tumor dose achieved is in the range of 72–100 Gy. Future work is needed to clarify which strategies can overcome biological tumor resistance and determine how best to coordinate systemic therapy integration with ablative dose regimens for patients with upfront unresectable PDAC.

-
Key concepts in vaccine therapy for PDAC
The application of vaccines as anticancer therapies dates to 1893, when William Coley, a surgeon in New York’s Memorial Hospital, observed tumor regression in patients injected with bacterial toxins (82). Advances in molecular biology and recombinant DNA technology have enabled the identification of TAAs and TSAs, transforming cancer vaccines into a personalizable therapeutic modality. By the late 20th century, investigators began exploring peptide-based, whole-cell, and dendritic cell (DC) vaccines, aiming to activate the immune system against tumor-specific targets (83). In PDAC, interest in vaccine therapy intensified with the identification of shared tumor antigens like KRAS, MUC1, and WT1 (84–86). Unfortunately, phase III studies in vaccine therapies for unresectable PDAC have failed to translate immune responses into improved clinical outcomes (87–89).
PDAC employs redundant mechanisms to escape immune detection and elimination, including downregulating antigen presentation and suppressing T cell activity through the recruitment of Tregs, myeloid-derived suppressor cells, and tumor-associated macrophages (78). This process often goes hand in hand with immune editing, a dynamic mechanism in which immunogenic tumor clones are selectively eliminated, leaving behind resistant variants (90). The low rate of tumor mutations in PDAC reduces the chances that an adaptive immune response will be induced owing to poor antigenicity (91).
Priming antigen-specific immune responses is critical to overcome the immunosuppressive nature of PDAC. DNA, RNA, and peptide vaccines are designed to present TAAs and TSAs to the host immune system, enabling the activation of CD4+ helper T cells and CD8+ cytotoxic T cells. These vaccines encode antigenic material translated or processed within the host, leading to antigen presentation through MHC class I and II pathways. This cross-presentation mechanism is essential for robust CD8+ T cell activation while supporting CD4+ T cell priming, which is critical for sustained immune responses and immune memory (Figure 1). DC-based vaccines involve the ex vivo differentiation and activation of autologous monocytes into highly effective antigen-presenting cells. These DCs are loaded with target peptides or tumor antigens in vitro, enabling them to prime and activate T cells upon infusion into the patient.
 Figure 1
Figure 1Mechanism of vaccine-induced cancer-specific immune response. The adjuvant or minimal residual disease setting represents an attractive approach for vaccine therapy in PDAC. Advantages include avoiding significantly higher tumor burden and its associated immunosuppression in the advanced/metastatic setting as well as optimizing the ratio of effector T cells to tumor cells.
Adjuvants play a pivotal role in enhancing antigenicity of vaccine strategies. Toll-like receptor (TLR) agonists, such as CpG oligonucleotides, mimic pathogen-associated molecular patterns to stimulate innate immunity and enhance T cell priming, and low-dose cyclophosphamide has been adapted to deplete Tregs. Thus, the integration of adjuvants into cancer vaccine strategies can be critical to achieve a robust and sustained antitumor immune response (92–95). A review of the literature offers perspective into the measurable immunogenicity of reported adjuvant vaccine platforms, permitting an assessment of therapeutic potential (Table 3).
Shared-antigen vaccines focus on targeting antigens overexpressed across cancers, including PDAC. For example, mutated KRAS is found in over 90% of PDAC cases and is a prime target for peptide-based vaccines like ELI-002 (96). Similarly, MUC1, an aberrantly glycosylated glycoprotein, and WT1, a transcription factor overexpressed in PDAC, have been incorporated into vaccine platforms such as GVAX and DC-based therapies. In contrast, neoantigen vaccines leverage the unique mutation-derived antigens of individual tumors. Platforms like autogene cevumeran use mRNA technology to deliver these personalized neoantigens, eliciting potent and tumor-specific T cell responses (97). The selection of neoantigens involves identifying mutation-derived epitopes with high affinity for a patient’s MHC molecules. Advances in next-generation sequencing and bioinformatics have greatly facilitated this process, enabling the rapid identification of immunogenic targets from tumor samples (97, 98).


Article tools
- Download citation information
- Send a comment
- Terms of use
- Standard abbreviations
- Need help? Email the journal
Review Series
Pancreatic Cancer
-
Pancreatic ductal adenocarcinoma: the Everest of cancer biology
-
Early neoplastic lesions of the pancreas: initiation, progression, and opportunities for precancer interception
-
What’s on the menu?: metabolic constraints in the pancreatic tumor microenvironment
-
Metastatic heterogeneity in pancreatic cancer: mechanisms and opportunities for targeted intervention
-
Evolving concepts in adjuvant/neoadjuvant therapy for resectable pancreas cancer
Metrics
Go to
- Top
- Abstract
- Introduction
- Neoadjuvant versus adjuvant therapy for resectable PDAC
- Radiation therapy for resectable PDAC
- Radiation therapy for borderline or locally advanced PDAC
- Cancer vaccines as adjuvant PDAC therapy
- Key concepts in vaccine therapy for PDAC
- Pancreas cancer vaccine therapy for patients with minimal residual disease
- Challenges and future directions in adjuvant PDAC vaccines
- Conclusions
- Footnotes
- References
- Version history



Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)