Advertisement
Research ArticleGeneticsNeuroscience
Open Access | ![]() 10.1172/JCI182088
10.1172/JCI182088
TDP-43 dysregulation of polyadenylation site selection is a defining feature of RNA misprocessing in amyotrophic lateral sclerosis and frontotemporal dementia
Frederick J. Arnold,1 Ya Cui,2 Sebastian Michels,1,3 Michael R. Colwin,1 Cameron M. Stockford,1 Wenbin Ye,2 Vidhya Maheswari Jawahar,4 Karen Jansen-West,4 Julien Philippe,1 Ravinder Gulia,1 Yunzi Gou,1 Oliver H. Tam,5,6 Sneha Menon,1 Wendy G. Situ,1 Saira L. Cazarez,1 Aryan Zandi,1 Kean C.K. Ehsani,1 Sierra Howard,1 Dennis W. Dickson,4,7 Molly Gale Hammell,5,6 Mercedes Prudencio,4,7 Leonard Petrucelli,4,7 Wei Li,2 and Albert R. La Spada1,2,8,9,10
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Arnold, F. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by
Cui, Y.
in:
PubMed
|
Google Scholar
|

1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Michels, S. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Colwin, M. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by
Stockford, C.
in:
PubMed
|
Google Scholar
|
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Ye, W. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Maheswari Jawahar, V. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by
Jansen-West, K.
in:
PubMed
|
Google Scholar
|
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Philippe, J. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Gulia, R. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Gou, Y. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Tam, O. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Menon, S. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Situ, W. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Cazarez, S. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Zandi, A. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Ehsani, K. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Howard, S. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by
Dickson, D.
in:
PubMed
|
Google Scholar
|
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Gale Hammell, M. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by
Prudencio, M.
in:
PubMed
|
Google Scholar
|
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Petrucelli, L. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by Li, W. in: PubMed | Google Scholar
1Department of Pathology & Laboratory Medicine and
2Department of Biological Chemistry, University of California, Irvine, Irvine, California, USA.
3Department of Neurology, University of Ulm, Ulm, Germany.
4Department of Neuroscience, Mayo Clinic, Jacksonville, Florida, USA.
5Institute for Systems Genetics, NYU Langone Health, New York, New York, USA.
6Department of Neuroscience & Physiology, NYU Grossman School of Medicine, New York, New York, USA.
7Neuroscience Graduate Program, Mayo Graduate School, Mayo Clinic College of Medicine, Jacksonville, Florida, USA.
8Department of Neurology,
9Department of Neurobiology & Behavior, and
10UCI Center for Neurotherapeutics, University of California, Irvine, Irvine, California, USA.
Address correspondence to: Albert R. La Spada, Pathology & Laboratory Medicine, Neurology, Biological Chemistry and Neurobiology & Behavior, UCI Center for Neurotherapeutics, University of California Irvine, Interdisciplinary Science and Enginering Building, Room 2044, 419 S. Circle View Dr., Irvine, California 92697, USA. Phone: 949.824.7407; Email: alaspada@uci.edu.
Authorship note: FJA, YC, and S Michels are co–first authors.
Find articles by La Spada, A. in: PubMed | Google Scholar
Published June 2, 2025 - More info
J Clin Invest. 2025;135(11):e182088. https://doi.org/10.1172/JCI182088.
© 2025 Arnold et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: April 16, 2024; Accepted: April 1, 2025

-
Results
TDP-43 depletion induces widespread APA in neuronal cells. To identify APA events regulated by TDP-43 in neuronal cells, we employed the dynamic analysis of poly(A) from an RNA-Seq (DaPars) tool to previously published RNA-Seq datasets (Figure 1A). While a number of tools have been developed to quantify APA from bulk RNA-Seq data (23), we selected DaPars (25), which identifies and quantifies de novo poly(A) site usage by applying a linear regression model to localized changes in RNA-Seq read density within the 3′ UTR. Given the established role of TDP-43 in regulating the splicing of unannotated “cryptic” exons, we hypothesized that “cryptic,” unannotated poly(A) sites may be similarly utilized upon TDP-43 depletion or mutation. DaPars calculates the percentage of distal poly(A) site usage index (PDUI) for each transcript, with the results averaged across replicates to determine ΔPDUI in cells where TDP-43 was knocked down or mutated in comparison with control conditions. A positive ΔPDUI denotes increased relative expression of transcripts with a longer 3′ UTR, while a negative ΔPDUI signifies increased relative expression of transcripts with a shorter 3′ UTR.
 Figure 1
Figure 1TDP-43 depletion induces widespread APA. (A) Overview of the study. We applied the dynamic analysis of poly(A) from an RNA-Seq (DaPars) tool to published RNA-Seq datasets to detect APA events resulting from TDP-43 depletion or mutation in immortalized cell lines, stem cell–derived neurons, and postmortem ALS/FTD neocortex. We then performed experimental and functional validation of select targets. Volcano plots depicting APA events in SH-SY5Y cells (B), SK-N-BE(2) cells (C), and i3Neurons (D) in which TDP-43 was knocked down via shRNA treatment. APA genes with FDR adj. P < 0.05 and ΔPDUI ≥ 0.1 are depicted in red, and APA genes with FDR adj. P < 0.05 and ΔPDUI ≤ –0.1 are depicted in blue. (E) Venn diagram illustrating the intersection of APA events across datasets. Hypergeometric distribution test was used to determine the statistical significance of overlapping genes between datasets: SH-SY5Y cells versus SK-N-BE(2) cells, P = 1.68 × 10–51; SH-SY5Y cells versus i3Neurons, P = 0.011; SK-N-BE(2) cells versus i3Neurons, P = 6.01 × 10–06.
Using published RNA-Seq data from SH-SY5Y cells, SK-N-BE(2) cells, and i3Neurons in which TDP-43 was depleted by shRNA knockdown (12), we identified hundreds of genes in which TDP-43 knockdown resulted in significant APA (FDR adjusted [adj] P < 0.05, |ΔPDUI| 0.1) (Figure 1, B–D, Tables 1, 2, and 3, and Supplemental Tables 1–3; supplemental material available online with this article;https://doi.org/10.1172/JCI182088DS1). APA genes exhibit both 3′ UTR lengthening and shortening upon TDP-43 knockdown; however, we observed increased use of distal poly(A) sites in 69.9% (460/658) of APA genes in SH-SY5Y cells, 59.6% (429/720) of APA genes in SK-N-BE(2) cells, and 78.1% (250/320) of APA genes in i3Neurons. Nearly 20% of APA events were shared between the closely related SH-SY5Y and SK-N-BE(2) neuroblastoma cell lines (Figure 1E), including 4/10 of the top APA genes by P value (Tables 1 and 2). Additionally, 16.3% (52/320) of APA genes in i3Neurons overlapped with at least 1 of the immortalized neuronal cell lines (Figure 1E), indicating that, while APA is known to be highly cell-type specific (17), many APA events regulated by TDP-43 are conserved across multiple neuronal cell types.
We also applied DaPars to previously published transcriptome data sets in which TDP-43 was depleted by siRNAs in SH-SY5Y cells (11) or in human embryonic stem cell–derived motor neurons (hESC-MNs) for 96 hours (9) (Supplemental Figure 1 and Supplemental Tables 4 and 5). As expected, we observed fewer APA changes in cells in which TDP-43 was knocked down for a shorter duration or incompletely (~60% in hESC-MNs). Both datasets do, however, exhibit significantly decreased PDUI in SMC1A, as previously shown for HEK293 cells upon TDP-43 siRNA knockdown (14).
Given that APA can affect cellular function by regulating mRNA stability and subcellular localization (22), we performed Gene Ontology (GO) analysis of APA genes in each neuronal cell type (Supplemental Figure 2). Shared terms between datasets for biological pathways known to be highly relevant to ALS/FTD included “establishment of protein localization” (SH-SY5Y and i3Neurons) and “cytoskeleton organization” (SH-SY5Y and SK-N-BE[2]), and shared terms for GO molecular functions were “kinase activity,” “protein kinase binding,” and “cytoskeletal protein binding” (SH-SY5Y and SK-N-BE[2]). These results indicate that TDP-43 regulation of APA impacts disease-relevant pathways and should be considered, along with differential expression and alternative splicing, as a key aspect of TDP-43 dysfunction.
TDP-43 binds within the 3′ UTR of a subset of APA genes, preferentially blocking the use of the distal PAS. By cross-referencing significant APA genes with TDP-43 enhanced crosslink and immunoprecipitation followed by sequencing (eCLIP-Seq) data from SH-SY5Y cells (26), we found that TDP-43 directly binds either within the 3′ UTR or in another region of the transcript to 43.2% (284/658) of APA genes in SH-SY5Y cells, 47.4% (341/720) of APA genes in SK-N-BE(2) cells, and 51.3% (164/320) of APA genes in i3Neurons (Figure 2A and Supplemental Figure 3). In neuronal cells chronically depleted of TDP-43, it is likely that cytotoxicity contributes to gene regulatory changes that do not necessarily reflect direct regulation by TDP-43. Indeed, we noted that in i3Neurons, only 36.7% of differentially expressed genes are targets of TDP-43 binding by eCLIP-Seq, while a higher proportion of CE (60.3%) and APA (51.3%) events are observed in genes directly bound by TDP-43 (Table 4). In support of our overall finding that TDP-43 depletion preferentially increases use of a distal PAS, we found that APA genes exhibit increased PDUI upon TDP-43 loss — when TDP-43 binds within the 3′ UTR, with i3Neurons displaying the largest trend (Figure 2, B–D).
 Figure 2
Figure 2TDP-43 binds within the 3′ UTR of a subset of APA genes, preferentially blocking use of the distal PAS. (A) Venn diagram illustrating the proportion of APA genes for which there is published evidence of TDP-43 binding within the 3′ UTR. (B–D) Graphical representation of ΔPDUI for all APA genes with TDP-43–binding sites in the 3′ UTR.
 Table 4
Table 4Percentage of DEGs, CE-containing genes, and APA genes with eCLIP-Seq–defined TDP-43 binding sites
To experimentally validate TDP-43 APA events, we first utilized the above SH-SY5Y cell model, in which a doxycycline-inducible shRNA against TDP-43 is stably integrated (12). We selected 2 APA genes to validate based on their robust and consistent shift in PDUI upon TDP-43 knockdown, and because TDP-43 binds directly within each 3′ UTR (Supplemental Figure 4). The Canopy FGF signaling regulator 3 (CNPY3) gene showed among the 3 greatest PDUI increases in each of our tested neuronal cell lines (Figure 2, B–D). CNPY3 exhibits extensive alternative splicing, with numerous annotated transcript variants. In examining the RNA-Seq tracks for CNPY3, we found that TDP-43 knockdown significantly increases PAS usage within a specific transcript variant (NM_001318848.2, variant 2), defined by an alternative last exon without an upstream splice junction. In line with these data, we confirmed by quantitative reverse transcription PCR (qRT-PCR) that TDP-43 depletion results in a significant increase in the use of the variant two 3′ UTR relative to variant one (NM_006586.5) in SH-SY5Y cells (Figure 3A and Supplemental Figure 5A). To demonstrate that CNPY3 APA is specifically regulated by TDP-43, we designed minigene expression constructs in which exon 3, intron 3, and exon 4 of the CNPY3 gene (NM_006586.5) were cloned downstream of nanoluciferase with either WT sequence or with the TDP-43–binding site mutated. We found that deletion of the TDP-43–binding motif was sufficient to recapitulate the CNPY3 isoform switch observed upon TDP-43 knockdown (Figure 3B). We further validated that CNPY3 APA occurs in i3Neurons following TDP-43 depletion (Figure 3C and Supplemental Figure 6A). Interestingly, we observed a cell-type–specific effect of TDP-43 knockdown on CNPY3 variant 1, whereby i3Neurons but not SH-SY5Y cells display a modest, but significant increase in CNPY3 variant 1 expression in addition to a substantial increase in variant 2 expression (Supplemental Figure 5). Because we found an overall increase in both CNPY3 isoforms in i3Neurons depleted of TDP-43, we evaluated CNPY3 protein levels. In contrast with increased expression of CNPY3 at the RNA level, we found that CNPY3 protein expression is decreased upon TDP-43 knockdown (Figure 3D). This suggests that TDP-43 regulation of CNPY3 APA may be functionally relevant in human neurons.
 Figure 3
Figure 3CNPY3 APA increases the expression of an isoform variant with an alternative last exon in neuronal cells and in ALS/FTD and FTLD-TDP patient tissue. (A) RT-PCR analysis and qRT-PCR quantification of CNPY3 APA isoforms with or without TDP-43 knockdown in SH-SY5Y cells. **P < 0.01; unpaired 2-tailed t test. n = 3 biological replicates. (B) RT-PCR analysis and qRT-PCR quantification of CNPY3 APA isoforms in SH-SY5Y cells transfected with minigene constructs encoding exon 3, intron 3, and exon 4 of the CNPY3 gene (NM_006586.5) with WT sequence or with the TDP-43–binding motif deleted (Del). *P < 0.05; unpaired 2-tailed t test. n = 4 biological replicates. (C) RT-PCR analysis and qRT-PCR quantification of CNPY3 APA isoforms with or without TDP-43 knockdown in i3Neurons. **P < 0.01; unpaired 2-tailed t test. n = 3 biological replicates. (D) Immunoblot analysis of CNPY3 in i3Neurons reveals that CNPY3 APA corresponds with a decrease in CNPY3 protein levels. *P < 0.05; unpaired 2-tailed t test. n = 3 biological replicates. (E) qRT-PCR quantification of CNPY3 APA isoforms in postmortem frontal cortex from healthy controls versus FTLD-TDP or ALS/FTD patients with confirmed CE inclusion in UNC13A. **P < 0.01; unpaired 2-tailed t test. n = 11 (control), n = 30 (UNC13A CE). All data are represented as mean values ± SEM.
Given that CNPY3 was one of the most consistent APA events observed across cell models (Figure 2, B–D), we next investigated CNPY3 APA in postmortem frontal cortex tissue from FTLD-TDP and ALS/FTD patients. Because even healthy controls exhibit low, but detectable levels of phosphorylated TDP-43 by ELISA, we used the presence or absence of the UNC13A CE as a more sensitive proxy for TDP-43 dysfunction (Table 5 and Supplemental Table 6). In agreement with our bioinformatics analyses and our experimental validation of CNPY3 APA in cell models of TDP-43 depletion, we observed significant CNPY3 APA, corresponding with increased expression of CNPY3 isoform variant 2 in the frontal cortex of TDP-43 proteinopathy patients (Figure 3E). Altogether, these results demonstrate that TDP-43 regulation of poly(A) site selection occurs in the CNS of ALS/FTD and FTLD-TDP patients. Specifically, CNPY3 APA increases the expression of a distinct protein-coding CNPY3 isoform with unknown function.
 Table 5
Table 5Clinical and molecular pathology of frontal cortex tissue from FTLD-TDP patients, ALS/FTD patients, and controls
In contrast to the majority of APA genes in which TDP-43 binds within the 3′ UTR, DaPars calculated a significant decrease in distal PAS usage for SMC1A (Supplemental Figure 4B), which we experimentally validated by qRT-PCR in SH-SY5Y cells (Figure 4, A and B). As noted, APA of SMC1A was previously characterized in HEK293 cells upon TDP-43 knockdown (14), indicating that this is a highly sensitive APA event across diverse cell types. Given that SMC1A is a subunit of the cohesin complex and plays a critical role in chromatin organization, we sought to investigate SMC1A in neuronal cells. TDP-43 depletion in i3Neurons resulted in significantly decreased expression of the long 3′ UTR SMC1A isoform and, in contrast to SH-SY5Y cells, increased expression of the short 3′ UTR SMC1A isoform (Figure 4, C and D). Furthermore, we found that SMC1A APA corresponded to a nearly 3-fold increase in SMC1A protein expression (Figure 4E). To determine whether TDP-43 regulation of SMC1A occurs in other neuronal cell types affected by TDP-43 proteinopathy, we similarly evaluated SMC1A APA and protein expression in DIV38 iPSC-derived motor neurons (iPSC-MNs) in which TDP-43 was knocked down for 10 days (Supplemental Figure 6B). We confirmed that TDP-43 loss indeed reduces use of the distal SMC1A PAS in human motor neurons (Figure 4, F and G). We further found that APA of SMC1A corresponds with a significant increase in SMC1A protein levels in iPSC-MNs (Figure 4H), suggesting that altered regulation of SMC1A APA may contribute to impaired chromatin organization upon TDP-43 loss, as previously reported in postmortem neuronal nuclei (27).
 Figure 4
Figure 4SMC1A APA corresponds with increased SMC1A protein levels in neuronal cells. (A, C, and F) RT-PCR analysis and (B, D, and G) qRT-PCR quantification of SMC1A APA in the presence or absence of TDP-43 knockdown in SH-SY5Y cells, i3Neurons, and iPSC-MNs, respectively. *P < 0.05; **P < 0.01; ***P < 0.001; unpaired 2-tailed t test. n = 3 biological replicates. (E and H) Immunoblot analysis of SMC1A reveals that SMC1A APA corresponds with an increase in SMC1A protein levels in i3Neurons and in iPSC-MNs, respectively. *P < 0.05; ***P < 0.001; unpaired 2-tailed t test. n = 3 biological replicates. All data are represented as mean values ± SEM.
Mutant TDP-43 induces APA in genes that function in the oxidative stress response. To explore APA dysregulation that reflects TDP-43 mutation in addition to TDP-43 knockdown, we next applied DaPars to RNA-Seq data generated from SH-SY5Y cells with homozygous mutation of TDP-43N352S achieved via CRISPR/Cas9 genome editing (11). Consistent with prior studies, 72% (59/82) of significant APA events corresponded with increased use of a distal PAS (Figure 5A, Supplemental Table 7). Notably, GO analysis revealed enrichment of genes that function in the “response to oxidative stress” pathway (Figure 5B). Numerous studies have implicated oxidative stress in ALS/FTD pathogenesis, including recent evidence that TDP-43 aggregation induces the generation of ROS (28). To determine whether TDP-43N352S SH-SY5Y cells display an altered oxidative stress response, we measured ROS at 30 minutes and 120 minutes after treating WT or TDP-43N352S SH-SY5Y cells with hydrogen peroxide. While there was no initial difference in ROS levels between control and TDP-43N352S cells, ROS levels were significantly higher in TDP-43N352S SH-SY5Y cells compared with WT cells after 120 minutes of exposure (Figure 5C). This provides proof of concept that TDP-43 APA genes function in cellular pathways implicated in neurodegenerative disease and that characterization of APA events can highlight disease-relevant phenotypes in cell models of ALS.
 Figure 5
Figure 5Mutant TDP-43 induces APA in genes functioning in oxidative stress response. (A) Volcano plot depicting APA genes in SH-SY5Y cells expressing homozygous TDP-43N352S via CRISPR/Cas9 mediated genome editing. APA genes with FDR adj. P < 0.05 and ΔPDUI ≥ 0.1 are depicted in red, and APA genes with FDR adj. P < 0.05 and ΔPDUI ≤ –0.1 are depicted in blue. (B) GO biological processes analysis of APA events in TDP-43N352S cells reveals impaired response to hydrogen peroxide (H2O2). (C) WT and TDP-43N352S cells were treated with 100 μM H2O2. ROS was detected at 30 minutes and 120 minutes using a fluorometric intracellular ROS detection kit. *P < 0.05, unpaired 2-tailed t test. n = 3 biological replicates. All data are represented as mean values ± SEM.
Nuclear clearance of TDP-43 induces APA in ALS/FTD patient neurons. To further determine the significance of TDP-43 APA dysregulation in human patients, we considered APA events in neuron nuclei obtained from 7 postmortem ALS/FTD neocortex samples, where FACS sorting resulted in transcriptome data sets for neurons either containing or depleted of nuclear TDP-43 (27). We identified 87 APA genes (|ΔPDUI| > 0.1, P < 0.05) in neuronal nuclei lacking nuclear TDP-43 (Figure 6A, Table 6, and Supplemental Table 8), but unlike in neuronal cell culture models, we observed a preference toward negative ΔPDUI in postmortem TDP-43 nuclear-depleted neurons, as 72.7% (63/87) of genes exhibited 3′ UTR shortening. By correlating the most significant APA events (|ΔPDUI| > 0.1, FDR P < 0.05) with eCLIP-Seq of TDP-43, we noted that 57.7% of these APA genes are bound by TDP-43 (Table 7), suggesting that many APA events are directly regulated by TDP-43. We then performed GO analysis for APA events in coding mRNA with P < 0.05 and [|ΔPDUI| ≥ 0.1] and found enrichment of genes functioning in the “histamine response,” “synapse assembly,” and “protein transport” pathways (Supplemental Figure 7). These pathways have been previously implicated in ALS disease models, again underscoring potential contributions of APA events to ALS pathobiology (29–31).
 Figure 6
Figure 6Nuclear clearance of TDP-43 induces APA in ALS/FTD patient neurons. (A) Volcano plot depicting APA genes in neuronal nuclei from 7 postmortem ALS/FTD neocortex samples sorted by FACS for the presence or absence of nuclear TDP-43. APA genes with P < 0.05 and ΔPDUI ≤ –0.1 are depicted in blue and APA genes with P < 0.05 and ΔPDUI ≥ 0.1 are in red. (B) eCLIP-Seq data showing the location of TDP-43 binding within the MARK3 3′ UTR, as well as in an upstream intronic region (green). MARK3 binding within the 3′ UTR is immediately upstream of the distal shift in 3′ UTR usage observed in TDP-43 negative neurons (blue versus red RNA-Seq tracks). (C) RT-PCR analysis and qRT-PCR quantification of MARK3 APA in SH-SY5Y cells transfected with minigene constructs in which the MARK3 3′ UTR was cloned downstream of the NanoLuc luciferase gene with WT sequence or with the TDP-43 binding motif deleted (Del). *P < 0.05; unpaired 2-tailed t test. n = 4 biological replicates. (D) RT-PCR of distal 3′ UTR (top band) and proximal 3′ UTR (bottom band) of MARK3 in iPSC-MNs in which TDP-43 was knocked down with shRNA for 10 days. *P < 0.05; unpaired 2-tailed t test. n = 3 biological replicates. All data are represented as mean values ± SEM.
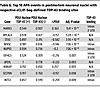 Table 6
Table 6Top 10 APA events in postmortem neuronal nuclei with respective eCLIP-Seq–defined TDP-43 binding sites
 Table 7
Table 7Percentage of CE-containing genes and APA genes with eCLIP-Seq–defined TDP-43–binding sites in ALS/FTD neuronal nuclei depleted of TDP-43
The most substantial 3′ UTR lengthening in ALS/FTD neuronal nuclei (ΔPDUI = +0.363), was observed in the gene encoding MARK3 (Table 6). Visualizing RNA-Seq tracks from this experiment with overlaid eCLIP-Seq generated by the ENCODE project (32), we confirmed that TDP-43 binds MARK3 in its 3′ UTR immediately upstream of a canonical ATTAAA poly(A) signal (hg38: chr14:103503803-103503809) as well as in an upstream intron (Figure 6B). This suggests that nuclear TDP-43 normally represses use of a distal MARK3 PAS, which then becomes preferentially utilized upon nuclear depletion of TDP-43. To test this hypothesis, we designed minigene expression constructs in which the MARK3 3′ UTR was cloned downstream of nanoluciferase with either WT sequence or with the TDP-43 binding site mutated. We found that deletion of the TDP-43–binding motif was sufficient to drive increased distal PAS usage in the MARK3 3′ UTR, supporting the hypothesis that TDP-43 represses use of the distal PAS (Figure 6C). Importantly, significant 3′ UTR lengthening in MARK3 was also observed in our APA analysis in SH-SY5Y and SK-N-BE(2) cells (Figure 2, B and C), indicating that increased utilization of a distal poly(A) site in MARK3 is a prominent effect in neuronal cells upon loss of nuclear TDP-43. Given the consistency of this result, we evaluated this phenotype in iPSC-MNs and confirmed that TDP-43 knockdown can induce markedly increased use of a distal PAS in the MARK3 gene (Figure 6D).
MARK3 APA promotes increased tau S262 phosphorylation and altered subcellular distribution in neurons. As MARK3 is a tau kinase associated with tau S262 phosphorylation in the early stages of AD pathogenesis (24), and because mutations in closely related MARK4 can significantly increase AD risk, promote hyperphosphorylation of tau, and induce neuron toxicity (33), we sought to investigate the functional consequences of MARK3 APA in neurons. qRT-PCR analysis revealed that TDP-43 knockdown results in increased use of the distal MARK3 PAS in i3Neurons (Figure 7A), which corresponds with an increase in overall MARK3 expression (Figure 7B). To characterize the cis-regulatory elements within the MARK3 3′ UTR that may affect its metabolism, we evaluated predicted miRNA-binding sites using miRBD (34) and TargetScan (35), and we obtained predicted RBP-binding motifs using RBPmap (36) (Supplemental Figure 8). Both miRNA prediction tools highlighted a conserved binding motif for miR-142-3p within the MARK3 3′ UTR (Supplemental Table 9). Intriguingly, this miRNA was found to be upregulated in ALS mouse models and in sporadic ALS patients; indeed, serum levels of miR-142-3p have been negatively correlated with ALS clinical outcomes (37). We also identified 2 conserved RBP-binding motifs within the MARK3 3′ UTR, which are only present when poly(A) occurs at a distal MARK3 PAS (Supplemental Figure 8 and Supplemental Table 10). The first motif is recognized by TDP-43 as well as by other RBPs (e.g., RBM24 and RBM38), while the second T/G-rich motif is predicted to be bound by several other RBPs, including TIA1 and HuR (Supplemental Figure 8B). The presence of highly conserved miRNA and RBP-binding motifs in the distal region of the MARK3 3′ UTR suggests that differential recognition of short versus long 3′ UTR isoforms by cis-regulatory elements may account for observed changes in MARK3 mRNA expression upon TDP-43 knockdown.
 Figure 7
Figure 7MARK3 APA corresponds with increased MARK3 transcript levels, increased tau S262 phosphorylation, and a change in MARK3 subcellular localization. (A) qRT-PCR quantification of MARK3 APA or (B) total MARK3 with or without TDP-43 knockdown in i3Neurons. *P < 0.05; **P < 0.01; unpaired 2-tailed t test. n = 3 biological replicates. (C) qRT-PCR quantification of total MARK3 expression in control (n = 52) or FTLD (n = 221) postmortem frontal cortex tissue. *P < 0.05; Mann-Whitney U test. (D) Immunoblot analysis of S262 phosphorylated tau and total tau in i3Neurons transduced with lentivirus encoding control shRNA or shRNA TARDBP for 10 days and treated with DMSO (vehicle) or with 5 μM of PCC0208017 (MARK3/4 inhibitor) for the final 24 hours. *P < 0.05; ***P < 0.001; 1-way ANOVA. n = 3 biological replicates. (E) Representative images of i3Neurons immunostained for MARK3 and total tau in the presence or absence of TDP-43 knockdown. Scale bars:10 μm. (F) Representative images of i3Neurons immunostained for tau pS262 and total tau in the presence or absence of TDP-43 knockdown. Scale bars: 10 μm. (G) Quantification of fluorescence intensity. 15–20 images were analyzed per condition, *P < 0.05; 1-sample t test. n = 3 biological replicates. All data are represented as mean values ± SEM.
Using a large cohort of frontal cortex tissue from healthy controls (n = 52) and FTLD-TDP patients (n = 221), we found that MARK3 RNA expression is modestly increased in the frontal cortex in human disease (Figure 7C and Supplemental Table 11). Using a smaller, independent cohort of patient tissue (Table 5 and Supplemental Table 6), we validated our finding of increased MARK3 RNA expression in the frontal cortex of ALS/FTD and FTLD-TDP patients relative to controls (Supplemental Figure 9, A and B). While we observed a small increase in the relative expression of the long 3′ UTR MARK3 isoform in TDP-43 proteinopathy patient tissue, this did not reach statistical significance (Supplemental Figure 9C). Given that MARK3 APA is prominent in monocultured neuronal cells (Figure 1, B and C) and in isolated neuronal nuclei depleted of TDP-43 (Figure 6A), but not in bulk frontal cortex tissue (Supplemental Figure 9C), it is likely that MARK3 RNA isoform expression is regulated by distinct mechanisms in nonneuronal cells or in neurons without TDP-43 pathology in FTLD-TDP.
Based on our finding that TDP-43 depletion results in an overall increase in MARK3 expression in i3Neurons and in postmortem patient tissue, we next evaluated tau S262 phosphorylation, a known target of MARK3 kinase activity. We found that TDP-43 knockdown significantly increases tau S262 phosphorylation in i3Neurons and that this effect is blocked by PCC0208017, a small molecule inhibitor of MARK3/4 (38) (Figure 7D). We similarly found that TDP-43 depletion in iPSC-MNs corresponds with a strong trend (P = 0.069) toward increased tau S262 phosphorylation (Supplemental Figure 10A). To determine whether MARK3 itself is sufficient to drive tau S262 hyperphosphorylation in human motor neurons, we transduced iPSC-MNs with lentivirus encoding a shRNA vector against MARK3, lentivirus encoding the MARK3 gene, or control empty vector lentivirus. In iPSC-MNs subjected to MARK3 overexpression, we detected an approximately 2.4-fold increase in tau S262 phosphorylation (Supplemental Figure 10B). Moreover, we observed an approximately 50% reduction in tau S262 phosphorylation in iPSC-MNs subjected to MARK3 shRNA knockdown (Supplemental Figure 10B).
While these results demonstrate that MARK3 protein levels correlate with tau S262 phosphorylation in iPSC-MNs and that inhibition of MARK3/4 kinase activity blocks increased tau S262 phosphorylation in i3Neurons, we found no significant change in MARK3 protein expression in i3Neurons or iPSC-MNs upon TDP-43 knockdown (Supplemental Figure 10C and Supplemental Figure 11A). Similarly, MARK3 protein levels were unchanged in ALS/FTD and FTLD-TDP patient tissue relative to controls (Supplemental Figure 11B). We further confirmed that protein levels of the closely related tau S262 kinase MARK4 are unaffected by TDP-43 knockdown (Supplemental Figure 11C). Given that we observed increased tau S262 phosphorylation upon TDP-43 knockdown, which was blocked by treatment with a MARK3/4 inhibitor, yet we observed no increase in MARK3 protein levels, we next examined whether MARK3 subcellular localization is altered by TDP-43 depletion in i3Neurons. Indeed, we noted a significant increase in the subcellular localization of MARK3 to neurites relative to soma upon TDP-43 knockdown, which corresponded with a similar trend of increased neurite localization of tau pS262 (Figure 7, E–G). Moreover, we observed a decrease in total tau localization in the neurites of i3Neurons upon TDP-43 depletion (Figure 7, E–G), suggesting broader dysregulation of tau dynamics following TDP-43 knockdown. Taken together, these results reveal a potentially important mechanistic link between TDP-43 and tau biology and suggest that TDP-43 dysregulation of neuronal MARK3 APA may contribute to altered cytoskeletal function in ALS/FTD and related neurodegenerative disorders.

















Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)