Advertisement
Research ArticleGeneticsInfectious disease
Open Access | ![]() 10.1172/JCI179822
10.1172/JCI179822
Myeloid cell genome-wide screen identifies variants associated with Mycobacterium tuberculosis–induced cytokine transcriptional responses
Joshua J. Ivie,1 Kimberly A. Dill-McFarland,2 Jason D. Simmons,2 Glenna J. Peterson,1 Penelope H. Benchek,3 Harriet Mayanja-Kizza,4 Lily E. Veith,2 Moeko Agata,2 Dang T.M. Ha,5 Ho D.T. Nghia,6,7,8 W. Henry Boom,9 Catherine M. Stein,3,9 Chiea C. Khor,10,11,12 Guy E. Thwaites,6,13 Hoang T. Hai,6,13 Nguyen T.T. Thuong,6,13 Xuling Chang,14 Sarah J. Dunstan,14 and Thomas R. Hawn2
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Ivie, J. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Dill-McFarland, K. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by
Simmons, J.
in:
PubMed
|
Google Scholar
|

1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Peterson, G. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Benchek, P. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Mayanja-Kizza, H. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Veith, L. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Agata, M. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Ha, D. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Nghia, H. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Boom, W. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Stein, C. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Khor, C. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Thwaites, G. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Hai, H. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Thuong, N. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Chang, X. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Dunstan, S. in: PubMed | Google Scholar
1Department of Global Health and
2Department of Medicine, University of Washington, Seattle, Washington, USA.
3Department of Population and Quantitative Health Sciences, Case Western Reserve University, Cleveland, Ohio, USA.
4Department of Medicine, School of Medicine, Makerere University, Kampala, Uganda.
5Pham Ngoc Thanh Hospital, Ho Chi Minh City, Vietnam.
6Oxford University Clinical Research Unit, Ho Chi Minh City, Vietnam.
7Hospital for Tropical Diseases, Ho Chi Minh City, Vietnam.
8Pham Ngoc Thach University of Medicine, Ho Chi Minh City, Vietnam.
9Department of Medicine, Case Western Reserve University, Cleveland, Ohio, USA.
10Genome Institute of Singapore, A-STAR, Singapore.
11Singapore Eye Research Institute, Singapore National Eye Centre and Eye ACP, Duke–National University of Singapore, Singapore.
12Department of Biochemistry, National University of Singapore, Singapore.
13Centre for Tropical Medicine and Global Health, Nuffield Department of Medicine, University of Oxford, Oxford, United Kingdom.
14Department of Infectious Diseases, University of Melbourne at the Peter Doherty Institute for Infection and Immunity, Parkville, Victoria, Australia.
Address correspondence to: Joshua J. Ivie, Seattle Children’s Research Institute, 1900 9th Avenue, Seattle, Washington 98101, USA. Phone: 206.884.3239; Email: jivie@uw.edu. Or to: Thomas R. Hawn, Department of Medicine, University of Washington, 750 Republican Street Box 358061, Seattle, Washington 98195-8061, USA. Phone: 206.616.4124; Email: thawn@uw.edu.
Find articles by Hawn, T. in: PubMed | Google Scholar
Published May 22, 2025 - More info
J Clin Invest. 2025;135(14):e179822. https://doi.org/10.1172/JCI179822.
© 2025 Ivie et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: January 31, 2024; Accepted: May 14, 2025
-
Results
Mtb-induced cytokine GWAS yields 77 genomic loci with suggestive associations. To discover genetic variants associated with differential Mtb-induced cytokine production, we examined 100 individuals in a Ugandan cohort with a previously generated RNA-Seq dataset of CD14+ monocytes infected for 6 hours with H37Rv Mtb compared with media control (Figure 1A and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI179822DS1) (41, 42). We examined IL1B, IL6, TNF, and IFNB1 log2 fold change in expression levels in this dataset, which were all highly increased during Mtb infection (Supplemental Figure 1A). All 100 individuals were simultaneously genotyped and imputed, yielding 8.3 million testable SNPs. We assessed each SNP using a linear model in the GENESIS package in R, adjusting for sex, age, and batch, and controlled for ancestry and population structure by adjusting for genotypic principal components (PCs), PC1, PC2, and kinship (Supplemental Figure 2). We then clustered individual SNP results into independent genomic loci using linkage disequilibrium–based (LD-based) clumping to the most significant, lead SNP within the locus. We did not identify any signal at a genome-wide level of significance (P < 5 × 10–8). However, a total of 77 genomic loci reached suggestive significance level (P < 1 × 10–5), including 51 for Mtb-induced IL1B, 24 for IL6, 6 for TNF, and 6 for IFNB1 (Figure 1B). Of the 77 total loci, 10 were found to have a suggestive association for both Mtb-induced IL1B and IL6 simultaneously. The majority of SNPs were associated with decreased Mtb-induced cytokine expression (Supplemental Table 2). In post hoc analyses, lead SNPs of many of these loci were found to be additionally associated with baseline, media cytokine expression (Supplemental Table 2). Although assessed independently, Mtb-induced IL1B expression, IL6 expression, and TNF expression were highly correlated (R = 0.51–0.8), and SNPs with a suggestive association with one cytokine often had some association in the other two (Supplemental Figure 1, B and C). In summary, 77 genomic loci were identified as candidate genetic regulators of the Mtb-induced cytokine response in a Ugandan cohort.
 Figure 1
Figure 1GWAS revealed 77 distinct genomic loci suggestively associated with Mtb-induced cytokine induction. (A) Diagram of workflow for the cellular GWAS approach and subsequent validation and mechanistic investigation. HHC indicates household contact cohort and HD indicates healthy donor cohort. Created with BioRender (biorender.com). (B) Manhattan plot shows GWAS results of all 4 cytokines plotted for –log10(P values). Results were calculated with a linear mixed model in GENESIS to account for population structure, genetic relatedness, sex, age, and RNA-Seq batch (Supplemental Figure 2). Dashed horizontal line indicates suggestive threshold, P < 1 × 10–5.
Functional annotation of candidate loci reveals potential mechanism and history of phenotypic effect. To identify potential mechanisms of genetic effect, we mapped each of the 77 loci to proximal genes, assessed expression quantitative trait loci (eQTL) effects, predicted deleterious SNPs, and identified previous locus associations in the literature and GWAS catalog (Table 1 and Supplemental Table 2) (43, 44). Many of the mapped genes have previously been associated with cytokine regulation and Mtb response (45–53). We further assessed potential cis-eQTL effects of loci SNPs on baseline, media expression of adjacent genes within 250 kb and identified several SNPs significantly associated with cis-eQTL effects (Supplemental Figure 3A). Additionally, many loci have SNPs with Combined Annotation Dependent Depletion (CADD) scores ranking them from the top 10% to 1% most deleterious SNPs (CADD = 10–20) (Supplemental Figure 3B). Lastly, multiple SNPs had previous COVID-19, autoimmune disease, protein QTL, and myeloid cell phenotype associations within the GWAS catalog. Together, these analyses demonstrate that many of the genomic loci identified have additional forms of evidence that support their potential to have an effect on Mtb-induced cytokine expression.
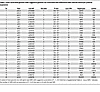 Table 1
Table 1Gene-associated genome-wide suggestive genomic loci associated with differential Mtb-induced monocyte cytokine expression
Gene set analysis: MAGMA identifies pathways enriched for Mtb-induced TNF effect. We performed gene set analysis (GSA) using Multi-marker Analysis of GenoMic Annotation (MAGMA), to determine whether SNPs had a significant joint association with Mtb-induced cytokine response in particular gene sets within the Molecular Signatures Database (MSigDB) (54–56). Gene P values were first determined from the aggregate signal of SNPs located within gene boundaries and adjusted for multiple correction while accounting for linkage disequilibrium (LD). GSA was then performed for each cytokine independently. Overall, across all gene sets and all 4 cytokines, only two significant associations were found, both of which were canonical pathways enriched for genes associated with Mtb-induced TNF (Figure 2A). One of these two significant pathways was α-linolenic acid metabolism, which has been implicated in direct effects on mycobacterial growth in addition to other macrophage response phenotypes (57–59). Analysis of the 19 gene-level P values and genotypic mean plots within the gene set identified early QQ plot deviation driven by numerous independent SNPs, indicating that significance marks a true gene set and multiple-SNP effect rather than being due to outlier effects (Supplemental Figures 4 and 5). Network visualization demonstrated that much of the α-linolenic acid metabolism pathway gene set is composed of a variety of PLA2 family members, which are known to regulate inflammation (60). The more significant gene-level P values appeared to be spread among multiple unrelated PLA2 isozymes within the pathway (Figure 2B). We next examined whether in vitro perturbation of PLA2 activity modulated the Mtb-induced cytokine response. Because of the previously identified correlation of IL1B, IL6, and TNF levels, we assessed the composite effect on these 3 inflammatory cytokines simultaneously. We used both live Mtb and Mtb whole-cell lysate (TBWCL) as stimuli to examine different activation pathways. We first compared the effects of the cytosolic PLA2 inhibitor AACOCF3 on macrophage cytokine production in response to TBWCL and identified a highly significant effect on Mtb-induced TNF secretion at the 10 μM dose (Figure 2C; P < 0.0001). No significant effect on IL1B or IL6 was observed. In contrast, upon live Mtb infection, we found that macrophage production of TNF was not significantly affected, whereas IL1B and IL6 were significantly decreased (Figure 2D; P < 0.001). In summary, these data showed that the aggregate effect of SNPs associated with Mtb-induced TNF expression was found to be significantly enriched in genes of the α-linolenic acid metabolism pathway. Further investigation of this gene set identified PLA2 as a major genetic regulator of Mtb-induced cytokine production, which was confirmed with cellular studies.
 Figure 2
Figure 2MAGMA enrichment identifies Mtb-induced TNF association with α-linolenic acid metabolism. (A) Previously calculated SNP P values for each cytokine were used to calculate gene-level P values, mapped to within each gene, accounting for SNP LD using MAGMA multi-model. Selected C2-canonical pathways (KEGG, Reactome, BioCarta, and Pathway Interaction Database) and C5-GO terms were assessed. Significant results after Bonferroni multiple correction are shown. Dashed vertical line indicates threshold for significance (adjusted P value < 0.05). (B) Network of genes within α-linolenic acid metabolism gene set was annotated for interaction using STRINGdb and plotted for MAGMA-calculated gene P value significance showing multiple PLA2 isoforms with increased significance. (C and D) TBWCL (C) and Mtb-induced secretion (D) of TNF, IL1B, and IL6 assessed by ELISA after increasing doses of cPLA2 inhibitor AACOCF3 pretreatment in human macrophages show that PLA2 enzymes do significantly affect TBWCL-induced TNF response and Mtb-induced IL1B and IL6 response. Results shown are from 3 biological replicates in 3 human donors per treatment. Significance was assessed using linear mixed model adjusting for random effect of donor followed by ANOVA and pairwise comparisons. Individual data points were plotted according to model calculated values adjusted for donor intercept. ***P < 0.001, ****P < 0.0001. Individual donor values are presented in Supplemental Figure 6A.
Multiple candidate loci have a population-spanning effect in an independent cohort. Since genetic effects can be population specific, we next examined whether any of the Uganda suggestive loci were associated with Mtb-induced cytokine production in an independent cohort. Using a cohort of 40 Seattle healthy donors, we stimulated monocyte-derived macrophages with Mtb or media alone and measured cytokine expression after 6 hours via quantitative PCR. The lead SNP of each locus suggestively associated with Mtb-induced cytokine expression in the Uganda cohort was examined in the Seattle cohort for association with Mtb-induced cytokine expression. Of the 77 SNPs in the original Uganda population, over half were unavailable to assess because of differences in SNP imputation and allele frequency between the Uganda and Seattle cohorts. However, of the remaining SNPs, 3 SNPs (rs10974620, rs79380271, and rs11881732) were found to be significantly associated with Mtb-induced IL1B expression and 1 SNP (rs16947739) significantly associated with Mtb-induced TNF expression at a nominal, P < 0.05 threshold (Supplemental Table 3). Because of differences in the number of loci tested per cytokine, only the SNP for Mtb-induced TNF (rs16947739) surpassed the significance threshold for multiple correction of number of SNPs tested. To account for differences between populations, all SNPs mapping within the 4 associated loci were assessed for LD and association with cytokine expression. Two of the 4 loci, located at chromosome (chr) 9p24.2 and chr 5q34, showed a number of SNPs in LD that were significantly associated with reduced Mtb-induced IL1B in both populations (Figure 3, A–D, and Supplemental Figure 7). The lead SNPs of the 2 loci (chr 9p24.2 and chr 5q34) were different in the 2 populations; rs10974620 and rs10815020 are the sentinel SNPs for the chr 9p24.2 loci, and rs79380271 and rs62378623 are the sentinel SNPs for the chr 5q34 loci, for Uganda and Seattle, respectively. The sentinel SNPs for Uganda are present in the majority of populations globally, with the exception of some parts of Asia for rs79380271 (Figure 3, E and F). Functional annotation revealed that the sentinel SNPs for the chr 9p24.2 and chr 5q34 loci were located within introns of the genes SLC1A1 and SLIT3, which have previously been linked to cytokine regulation in the literature (47, 48). Together, these data suggest that SLIT3 and SLC1A1 are lead candidate genes as major regulators of Mtb-induced IL1B in humans.
 Figure 3
Figure 3Multiple SNPs associated with Mtb-induced cytokine expression in the Uganda cohort show association with cytokine expression in a Seattle population cohort. Lead SNPs that passed the suggestive threshold for a particular cytokine within the Uganda population (n = 100) were tested for association with the corresponding cytokine in the Seattle population (n = 40). Four loci were associated with cytokine expression in the 2 populations (P < 0.05). To account for differences between populations, all SNPs within the 2 loci were assessed for linkage disequilibrium (LD) and association with corresponding cytokine expression. (A) P values of SNPs of the chr 9p24.2 locus, within the gene SLC1A1, with coloration of individual SNP points indicating LD with rs10974620 within each population. (B) Differences in Mtb-induced IL1B expression are plotted according to genotype for the chr 9p24.2 locus lead SNP of the Uganda population, rs10974620. (C) P values of SNPs of the chr 5q34 locus, within the gene SLIT3, with coloration of individual SNP points indicating LD with rs79380271 within each population. (D) Differences in Mtb-induced IL1B expression are plotted according to genotype for the chr 5q34 locus lead SNP of the Uganda population, rs79380271. Threshold lines indicate nominal P value threshold (blue, P < 0.05 unadjusted) and multiple corrected threshold (red, 0.05/number of SNPs tested for each cytokine). For both loci, multiple SNPs are significantly associated with decreased Mtb-induced IL1B expression in both populations. (E and F) Global minor allele frequency of the SLC1A1 locus Uganda sentinel SNP, rs10974620 (E), and SLIT3 locus Uganda sentinel SNP, rs79380271 (F), plotted using Geography of Genetic Variants browser.
SLC1A1 and SLIT3 affect Mtb-induced cytokine induction. To experimentally validate whether the identified candidate genes had an effect in vitro, we used siRNA in monocyte-derived macrophages (MDMs) and examined the effects of SLC1A1 and SLIT3 on Mtb-induced cytokine induction. Three or more siRNA sequences per gene were used to select an optimal targeting siRNA resulting in a more than 70% decrease in target gene expression (Supplemental Figure 8). RNAi knockdown of SLC1A1 significantly increased and SLIT3 significantly decreased Mtb-induced IL1B protein secretion and mRNA expression robustly across multiple human donors and siRNA target sequence (Figure 4, A and B, and Supplemental Figure 9; P < 0.01). SLIT3 knockdown also resulted in a highly significant decrease in IL1B expression in the media-only condition (Figure 4B; P < 0.0001). To assess a greater spectrum of inflammatory cytokines, we further assessed for the effect of IL6 and TNF expression. SLIT3 knockdown was associated with decreased Mtb-induced IL6 and TNF protein secretion (Figure 4A; P < 0.05) as well as mock-infected TNF mRNA expression (Figure 4B; P < 0.01). In summary, these in vitro data indicate that expression of the candidate genes SLC1A1 and SLIT3 is associated with Mtb-induced cytokine responses in macrophages.
 Figure 4
Figure 4In vitro investigation of SNP-associated genes identifies SLC1A1 and SLIT3 as major regulators of the Mtb-induced cytokine response. (A and B) siRNA knockdown of SLC1A1 and SLIT3 expression was used to determine effect on IL1B, IL6, and TNF secretion measured by ELISA after 24 hours of Mtb infection (MOI = 1) (A) and IL1B, IL6, and TNF mRNA expression after 6-hour mock and Mtb infection (B). (C) Purified hSLIT3 N- and C-terminal fragments were tested for effect on IL1B, IL6, and TNF secretion measured by ELISA after 24-hour mock and Mtb infection. Results are from 3 biological replicates in 3 human donors per siRNA or treatment. ND, not detected. Significance was assessed using linear mixed model adjusting for random effect of donor followed by ANOVA and pairwise comparisons. Individual data points were plotted according to model calculated values adjusted for donor intercept. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Individual donor values are presented in Supplemental Figure 6, B–D.
Exogenous application of N- and C-terminal human SLIT3 increases the Mtb-induced cytokine response. We next assessed whether extracellular application of purified SLIT3 regulated cytokine induction. SLIT3 is cleaved into an N-terminal and a C-terminal component, which bind to the ROBO and PLEXIN A receptors, respectively (61–64). Two of the four ROBO family receptors, ROBO1 and ROBO3, as well as four PLEXIN A family receptors were found to be expressed in human monocytes (Supplemental Figure 10). When N- and C-terminal SLIT3 were coapplied, Mtb-induced IL1B and TNF protein secretion was significantly increased (Figure 4C; P < 0.05). For Mtb-induced TNF, a significant effect is seen with N-SLIT3 pretreatment alone, and this was amplified when both fragments were applied. In the uninfected conditions, SLIT3 pretreatment induced IL6 and TNF but not IL1B secretion, with an effect that was largest with cotreatment of both the N- and C-terminal fragments (Figure 4C; P < 0.0001). However, in contrast to Mtb-induced TNF, only the C-SLIT3 fragment led to a significant increase in IL6 secretion baseline, with the N-terminal having no significant effect. Taken together, these results indicate that receptors for both the N- and C-terminal of SLIT3 are likely to be present in human MDMs and that N- and C-terminal SLIT3 treatment creates distinct effects on the cytokine response, which are amplified when they are applied simultaneously.
SLIT3 N- and C-terminal fragments induce increase in Mtb replication in MDMs. We next assessed whether SLIT3 had an effect on other Mtb-response macrophage phenotypes, including phagocytosis and intracellular replication. Using a microscopy-based assay measuring surface area of fluorescently labeled Mtb, we quantified relative abundance of macrophage-associated Mtb at 4 hours after infection as well as growth over a 72-hour period. No significant differences in phagocytic uptake were seen between any SLIT3 treatment and vehicle-treated control after 4 hours (Figure 5). However, the Mtb fluorescence surface area was increased in the N- and C-terminal–cotreated samples at both the 48- and 72-hour time points (Figure 5; P < 0.05). The N-terminal–only pretreatment had a trending but non-significant effect on growth at 72 hours, whereas the C-terminal–only pretreatment had no significant effect on Mtb growth at any time point. Overall, these results reveal that coapplication of SLIT3 N- and C-terminal fragments has a significant effect on intracellular replication of Mtb that is not due to initial differences in phagocytic uptake.
 Figure 5
Figure 5Evaluation of SNP-associated gene effect on other Mtb-response phenotypes reveals SLIT3 effect on intracellular Mtb. Intracellular Mtb replication in human macrophages assessed after hSLIT3 N- and C-terminal cotreatment using fluorescence microscopy to measure Mtb-mCherry surface area within each well over 72 hours. Four-hour time point quantifies differences in initial phagocytic uptake and shows no significant differences between groups. Results shown are from 6 biological replicates in 3 human donors. Significant differences were evaluated at each time point and show a significant increase in intracellular Mtb replication with cotreatment of N- and C-terminal SLIT3 at 48 and 72 hours. Significance was assessed using a linear mixed model adjusting for random effect of donor followed by ANOVA and pairwise comparisons for each time point. Individual data points were plotted according to model calculated values adjusted for donor intercept. *P < 0.05. Individual donor values are presented in Supplemental Figure 6E.
Assessment of candidate genomic loci mechanism of effect. Since the candidate functional genes, SLC1A1 and SLIT3, showed a validated effect on Mtb-induced cytokine induction, we further sought to identify the causal genetic mechanisms of their corresponding loci. The lead SNP of the SLC1A1 locus, rs10974620, was previously identified as an eQTL in lymphoblastoid cell lines (P < 1 × 10–5) (65). Within the iMETHYL database, which assesses the immune cell methylation landscape, rs10974620 was located at a highly methylated site in CD14+ monocytes and had significant methylation quantitative trait loci (mQTL) associations with numerous adjacent probes (P < 1 × 10–10) (66). In the Uganda cohort, the uncommon allele of rs10974620 was associated with increased levels of SLC1A1 expression in the Mtb but not the media condition (Supplemental Figure 11A; P < 0.01). To validate the iMETHYL findings, we used previously published global methylation profiling of CD14+ monocyte samples from our Ugandan cohort (67). Using the closest proximal methylation probe shared with the iMETHYL database, we validated a significant decrease in methylation according to rs10974620 allele frequency (Supplemental Figure 11B; P < 1 × 10–8).
For SLIT3, the lead SNP rs79380271 was not associated with eQTL effects. To determine whether there were protein quantitative trait loci (pQTL) effects, we also assessed secreted and cell lysate levels of SLIT3 protein abundance via ELISA, in MDMs from human donors in our Seattle cohort with various rs79380271 genotypes after 24 hours of media or TBWCL stimulation. Although we did not detect secreted SLIT3, we consistently identified SLIT3 protein levels in MDM lysate, which were decreased by TBWCL stimulation (Supplemental Figure 10C; P < 0.05). Interestingly, we observed a suggestive decrease in SLIT3 protein levels after media and TBWCL stimulation according to rs79380271 uncommon allele frequency in a small sample set (n = 10, P = 0.17 and 0.19, respectively) (Supplemental Figure 11C). Together, these data suggest that these genomic loci may regulate Mtb-induced IL1B expression through causally linked differences in levels of SLC1A1 and SLIT3.
Association of genetic variants with clinical TB phenotypes. Since the Mtb-induced cytokine response plays diverse roles in modulating the spectrum of clinical TB progression, we next explored whether our SLC1A1 and SLIT3 loci were associated with clinical TB phenotypes. We assessed 4 clinical phenotypes ranging in severity, including tuberculin skin test/IFN-γ release assay (TST/IGRA) conversion, pulmonary TB (PTB), tuberculous meningitis (TBM), and TBM survival. Association with TST/IGRA conversion was tested using the SLC1A1 lead SNP (rs10974620) and the SLIT3 lead SNP (rs79380271) in a previously characterized Ugandan household contact cohort (9). PTB, TBM, and TBM survival association was similarly tested in a previously characterized Vietnam cohort (68–71). Owing to population differences, the rs79380271 genotype was not present in the Vietnam dataset, and the SLIT3 Seattle lead SNP (rs62378623) was used as a proxy. Although no significant association was found for TST/IGRA conversion or PTB, both loci had SNPs associated with TBM phenotypes. The SLC1A1 SNP, rs10974620, was associated with higher susceptibility to TBM (odds ratio, 1.238; 95% CI, 1.037–1.478; P = 0.018) (Table 2). Additionally, TBM mortality was higher among individuals with the SLIT3 SNP, rs62378623, T/C genotype (6/9, 66.6%) versus the T/T genotype (57/360, 15.8%; hazard ratio, 23.63; 95% CI, 5.59–99.77; P = 1.05 × 10–4). Furthermore, evaluation of surrounding SNPs within each locus identified many SNPs in high LD that also showed significant association (Supplemental Figure 12). Together, these data indicate that the genomic loci within SLC1A1 and SLIT3 that are associated with Mtb-induced cytokine expression are also associated with the severe clinical TB phenotypes, TBM and TBM survival.










Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)