Advertisement
Review
Open Access | ![]() 10.1172/JCI205086
10.1172/JCI205086
Towards precision medicine for brain arteriovenous malformations
Andrew T. Hale,1,2 Adam J. Kundishora,3 Pazhanichamy Kalailingam,4 Tanyeri Barak,5 Phan Q. Duy,6 Christopher M. Ramundo,6 Baojian Fan,6 Qiang Li,6 Priscilla K. Brastianos,7 Ganesh M. Shankar,6 Seth L. Alper,7,8,9 Benjamin P. Kleinstiver,4,10,11 Patricia L. Musolino,4,12 and Kristopher T. Kahle6,9,13,14
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by
Hale, A.
in:
PubMed
|
Google Scholar
|

1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Kundishora, A. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Kalailingam, P. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Barak, T. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Duy, P. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Ramundo, C. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Fan, B. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Li, Q. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Brastianos, P. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Shankar, G. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Alper, S. in: PubMed | Google Scholar
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by
Kleinstiver, B.
in:
PubMed
|
Google Scholar
|
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by
Musolino, P.
in:
PubMed
|
Google Scholar
|
1Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
2Neuroscience Institute, University of Cape Town, Cape Town, South Africa.
3Division of Neurosurgery, Children’s Hospital of Philadelphia, Perelman School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania, USA.
4Center for Genomic Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
5Department of Neurosurgery, Yale University School of Medicine, New Haven, Connecticut, USA.
6Department of Neurosurgery, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
7Department of Medicine, Massachusetts General Hospital, Boston, Massachusetts, USA.
8Division of Nephrology and Vascular Biology Research Center, Beth Israel Deaconess Medical Center, Boston, Massachusetts, USA.
9Broad Institute of MIT and Harvard, Cambridge, Massachusetts, USA.
10Department of Pathology, Massachusetts General Hospital, Boston, Massachusetts, USA.
11Department of Pathology, Harvard Medical School, Boston, Massachusetts, USA.
12Department of Neurology, Harvard Medical School and Massachusetts General Hospital, Boston, Massachusetts, USA.
13Division of Genetics and Genomics, Boston Children’s Hospital, Boston, Massachusetts, USA.
14Harvard PhD Program in Neuroscience (PiN), Harvard University, Cambridge, Massachusetts, USA.
Address correspondence to: Andew T. Hale, 1720 2nd Ave. Birmingham, AL, 35233. Email: andrewthale20@gmail.com. Or to: Patricia L. Mulsolino, 55 Fruit St, Boston, MA 02114. Email: pmusolino@mgb.org. Or to: Kristopher T. Kahle, 15 Parkman St., WACC 331, Boston, MA 02114. Email: kahle.kristopher@mgh.harvard.edu.
Find articles by Kahle, K. in: PubMed | Google Scholar
Published May 15, 2026 - More info
J Clin Invest. 2026;136(10):e205086. https://doi.org/10.1172/JCI205086.
© 2026 Hale et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
-
Angiogenesis, AV specification, zonation, and CV development
CV growth, development, and remodeling occur through angiogenesis, either by sprouting from existing vessels or by partitioning into daughter branches via intussusception. These processes depend on tightly regulated molecular, genetic, and environmental cues (16). Both angiogenesis mechanisms involve proliferation, migration, patterning, cellular renewal, and morphological remodeling. Genetic and environmental modulators of angiogenesis — including shear stress (17), inflammation (18) and radiation (19) — shape CV identity, function, connections, and mechanical stability. Although multiple angiogenic phenotypes result from genetic alteration or pharmacologic manipulation of these pathways, we anticipate that specific knowledge of signaling pathways responsible for bAVM growth, maintenance, and regression will enable molecularly guided precision treatments. Aberrant spatial or temporal expression of angiogenic factors underlies bAVM histopathology (20), and risk variants in angiogenesis-associated genes may influence bAVM formation and hemorrhage (21). Therefore, framing molecularly guided bAVM treatment requires an overview of pathways regulating angiogenesis, AV specification, zonation, and CV development.
Ephrin/Eph/RASA1. The ten EphA receptor isoforms and six EphB receptor isoforms are receptor tyrosine kinases (RTKs) that coordinate bidirectional signaling through direct interactions with the five ephrin A ligand isoforms and three ephrin B isoforms to regulate angiogenesis (Figure 1) and other cellular processes (22). Exquisite control, crosstalk, and pleiotropy among members of the ephrin/Eph pathway control endothelial cell (EC) migration and adhesion and AV differentiation, specification, and zonation in normal and pathological CV (23). Expression of ephrin B2 and EphB4 define arterial and venous ECs, respectively, during development (24). This pattern is maintained (25) but the ratio of cell types is perturbed in human bAVMs (26). Whether this perturbation is a consequence of bAVM formation or a primary mechanistic driver of bAVM pathogenesis, growth, and/or maintenance is not known. Germline SNPs in EPHB4 have been associated with rupture risk in sporadic adult bAVMs (27), but the mechanistic basis for this observation is not clear. Our group and others have identified causative EPHB4 loss-of-function mutations in a subset of human patients with the congenital/pediatric bAVM subtype VOGM (14, 28, 29). Heterozygous mice expressing a VOGM-specific Ephb4 kinase domain missense variant (Ephb4F867L) displayed severe CV abnormalities only in the presence of a second variant allele or a Cre-disrupted Ephb4 allele (14).
 Figure 1
Figure 1Overview of ephrin/Eph/RASA1, PI3K/AKT/mTOR, VEGF, NOTCH, TGF-β/SMAD, BRAF, and KRAS pathways. These signaling pathways underlie angiogenesis, AV specification, and CV development. Eph, ephrin; ACVRL1/ALK1, activin receptor–like kinase 1; SMAD, suppressor of mothers against decapentaplegic; SARA, SMAD anchor for receptor activation; Smurf2, SMAD ubiquitin regulatory factor 2; VRAP, VEGF receptor–associated protein; Shc, Src-like kinase C; PTEN, phosphatase and tensin homolog deleted on chromosome 10; AKT, protein kinase B; mTORC1, mammalian target of rapamycin complex 1; PTPN1, tyrosine-protein phosphatase non-receptor type I; RASA1, p120 Ras GTPase–activating protein; RAF, rapidly accelerated fibrosarcoma.
Downstream activation of RAS suppressor p120 Ras GTPase–activating protein (RASA1), a negative regulator of RAS signaling, is the mechanism through which EphB4 mutations regulate angiogenesis. EphB4 and RASA1 physically interact via an SH2 domain in RASA1 and a phosphorylated tyrosine residue in EphB4 (30) to limit RAS signaling in ECs (14). RAS activation also causes downstream activation of VEGF-dependent signaling (31) and is sufficient to cause bAVMs in mice (32). Somatic RASA1 variants cause capillary malformation AVM syndrome (CM-AVM) (33, 34), characterized by high-flow vascular malformations, including systemic and intracranial AV fistulas, and, rarely, bAVM subtype VOGM (35, 36). Moreover, mice with an Ephb4 mutation that prevents physical interaction with RASA1 but retains protein tyrosine kinase activity show normal angiogenic phenotypes (37). These data suggest, but do not prove, that EphB4 and RASA1 act within the same pathway to regulate angiogenesis. Pharmacological manipulation of RASA1 or downstream activators (e.g., mTOR) may be an appealing approach to treat genetically susceptible bAVM subtypes. However, somatic activating mutations in EPHB4 and RASA1 have not been identified in human patients with sporadic bAVMs.
PI3K/AKT/mTOR. Phosphoinositide 3-kinases (PI3Ks) are lipid kinases (class I, II, and III) that control downstream signaling pathways, including the serine-threonine kinases AKT and mTOR to control cell growth, proliferation, migration, and survival in the context of angiogenesis (Figure 1). Ligand-receptor binding or environmental stimuli such as shear stress (17) promote phospholipase C–mediated cleavage of phosphoinositide 4,5-bisphosphate (PIP2) to generate the second messengers diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3) (38). Somatic gain-of-function mutations in PI3KCA have been identified in venous malformations (39) and cerebral cavernous malformations (40) in mice and humans, but not sporadic bAVMs or other bAVM subtypes. Pharmacologic inhibition of PI3K or mTOR leads to regression of PI3K-driven vascular malformations (41) and is therapeutic in mouse models of HHT (42, 43). However, in mice harboring EC-specific HrasG12V mutations, PI3K inhibition had no effect in vivo on AVM-like features (i.e., vascular dysplasia, hemorrhage, etc.) in the brain and other organs (32). Of note, to our knowledge, somatic activating mutations in HRAS, NRAS, or PI3K/AKT/mTOR pathway genes have not been identified in human patients with sporadic bAVMs. Importantly, inhibition of MEK, but not PI3K, signaling can abrogate angiogenesis through VEGF-dependent pathways in human KRAS-mutant bAVM ECs (10). Thus, pharmacotherapies against PI3K/AKT/mTOR are unlikely to provide therapeutic benefit for patients with sporadic bAVMs.
VEGF. The VEGFA-VEGFR2 interaction is the main signaling pathway driving CV angiogenesis, although other VEGF ligands (VEGFB–D) and receptors (VEGFR1–3) also have context-specific roles (Figure 1). VEGFA signaling has been extensively studied in bAVM pathogenesis in preclinical models, where overactivation of VEGFA causes CV dysmorphogenesis (44) and hemorrhage that can be attenuated by MMP9 inhibition (45). Additional support for VEGF signaling in bAVM pathogenesis is provided by mouse studies demonstrating that VEGF inhibition via treatment with the monoclonal antibody bevacizumab attenuates vascular dysplasia in mature bAVMs of Alk1-deficient mice (46). In contrast, two patients (genotype unknown) with sporadic bAVM treated with bevacizumab did not experience reduced bAVM volume, although the reduced VEGF serum levels observed at 26 weeks of treatment were not sustained through the end of the 52-week treatment period (47). Although no adverse systemic effects (hypertension, proteinuria, impaired wound healing, etc.) (48) of bevacizumab were reported in this case series, additional safety data are needed. In one series of patients with sporadic bAVMs, germline variants were identified in genes involved in VEGF and BMP/TGF-β pathways (49), but clear pathogenic evidence for these variants is lacking. Moreover, VEGF signaling can be augmented by shear stress (50) and iatrogenic bAVM embolization (51) through disruption of the blood-brain barrier (BBB) (52). These data highlight the highly dynamic regulation of VEGF in vivo, and suggest underlying mechanisms by which VEGF contributes to bAVM biology and treatment response. We hypothesize that VEGF signaling contributes to bAVM pathogenesis via augmentation of vascular stability and remodeling rather than as a primary genetic driver. While there is some compelling preclinical evidence, limited and variable human outcomes data, along with potential systemic toxicities, tempers enthusiasm for VEGF as a therapeutic target.
TGF-β/SMAD. TGF-β/SMAD signaling has both pro- and antiangiogenic roles. Clinical studies of HHT confirmed that loss-of-function variants in endoglin (ENG) (HHT1) or ALK1/ACVRL1 (HHT2), both members of the TGF-β superfamily, cause the disease (53, 54). For example, ALK1/ACVRL1 is a receptor that, when bound by a ligand, phosphorylates SMAD1/5/8, enabling the latter to form a complex with SMAD4 (mutated in HHT5) and translocate to the nucleus (Figure 1). Loss of ALK1/ACVRL1 removes the angiogenic “brake” on VEGF-mediated PI3K signaling, leading to abnormal sprouting, EC proliferation, and vascular dysplasia (55). Loss-of-function mutations in Eng, a transmembrane coreceptor for ALK1/ACVRL1, cause bAVMs through similar mechanisms (56–58). Mouse models lacking systemic or EC-specific Smad4 exhibit systemic AVMs and bAVMs that respond to angiopoietin-2 inhibition (59–63). Furthermore, decreased expression of TGF-β/BMP and downstream SMAD6 has been linked to microhemorrhage in unruptured human sporadic bAVM surgical samples (64). Thus, it appears that changes in TGF-β/SMAD signaling, whether directly or via upstream regulation, may contribute to the development of bAVM pathogenesis through complex regulation of angiogenesis.
Notch. Notch signaling occurs through ligand-receptor interactions (DLL1/3/4, JAG1/2 binding NOTCH1–4), resulting in γ-secretase–dependent release of the Notch intracellular domain (NICD) and nuclear translocation to regulate transcription via CBF-1/RBPJ (Figure 1). Modulation of NOTCH1, -3, and -4 and RBPJ — leading to either increased or decreased Notch signaling — has been implicated in bAVM, extracranial AVM, and vascular dysgenesis in mice (65–73), but no pathogenic NOTCH mutations have been identified in human sporadic bAVMs. Interpretation of Notch-dependent phenotypes is complicated by downstream signaling redundancy, crosstalk, varied transcriptional programs, cell-type specificity, sequestration of Notch binding partners, differential ligand binding, and compensatory mechanisms (74). For example, deletion of the downstream Notch effector gene Rbpj has been shown to both cause vascular dysmorphogenesis and also rescue NOTCH4-dependent AVM formation (71–73), whereas Notch deficiency in pericytes causes extracranial AVMs and bAVMs (75). Mice lacking EphB4 or Efnb2 feature embryonic vascular defects similar to those observed in mice harboring Notch1 gain-of-function mutations (67), but these phenotypes do not resemble sporadic, adult-onset bAVMs. Germline NOTCH4 variants have also been suggested to increase risk of human bAVM formation (65), but independent replication and direct causal evidence is lacking. Additional data support shear stress– and vascular tone–mediated regulation of Notch contributing to bAVM pathogenesis (76, 77), and the therapeutic effect of radiation on bAVMs may be mediated in part by Notch signaling (78). Although Notch perturbation in mice rarely results in bAVM phenotypes independently from angiogenic modulation, these studies have been instrumental in identifying the essential role of Notch signaling in vascular development. The overlapping and convergent mechanisms with ephrin and TGF-β/SMAD suggest that these pathways may be modulated in specific bAVM biological states and in response to radiation. Thus, the role of Notch in human sporadic bAVM pathogenesis is likely dependent on genotype and environment.
KRAS. KRAS, a GTPase downstream of RTKs, is a major cellular regulator of growth and proliferation and has been extensively studied in oncology. The discovery of somatic mutations in KRAS underlying approximately 50%–80% of sporadic bAVMs (8–10) has directed attention to the role of KRAS as an angiogenesis regulator (Figure 1). KRAS activation has been shown to augment endothelial-mesenchymal transition (endoMT) through upstream modulation of TGF-β/SMAD, through a mechanism attenuated by lovastatin (79). KRAS-activating mutations cause downstream activation of PI3K-independent MAPK/ERK signaling (80). The prevalence of KRAS gain-of-function mutations in sporadic bAVMs suggests that anti-VEGF therapy may be unsuccessful, although as noted above there are very limited bevacizumab bAVM treatment data in human patients (47). KRAS-mutant-specific pharmacology, downstream MEK inhibition, and primary correction of pathogenic somatic mutations (or KRAS hyperactivation) may be viable nonsurgical treatments for human sporadic bAVMs, but additional mechanistic studies coupled with rigorous clinical trials and safety data are needed.

-
Genetic basis of bAVM pathogenesis
Human genetics of syndromic bAVMs. Syndromes associated with bAVM development — caused by autosomal dominant inherited, or less commonly, de novo variants — include HHT1, -2, and -5 (caused by mutations in ENG, ALK1/ACVRL1, and SMAD4, respectively) (53, 54), CM-AVM (RASA1 and EPHB4 mutations) (13, 81), Parkes-Weber syndrome (RASA1 mutations) (35), and Sturge-Weber syndrome (somatic GNAQ mutations) (82) (Table 1). However, most cases are incompletely penetrant and variably expressive, leading to AVMs in different locations (brain, lung, liver, etc.), where somatic second-hit loss-of-function mutations or triggers (i.e., transzygosity in modifier genes, shear stress, angiogenic stimuli, etc.; ref. 83) have been shown to determine the spatiotemporal parameters of AVM pathogenesis. Accepted mechanisms causing bAVMs in HHT include changes in PI3K activation and aberrant Notch signaling (55, 84), whereas CM-AVM cases caused by EPHB4 and RASA1 mutations lead to aberrant activation of RAS signaling (13, 81). However, these bAVMs differ pathologically. HHT-associated bAVMs are typically lower-flow and characterized by vessel dysplasia (more common in patients with HHT1 [~20%] than in those with HHT2 [5%]), with systemic clinical signs (i.e., mucocutaneous capillary malformations) typically appearing in adolescence or adulthood. In contrast, CM-AVM–related brain and other AVMs are high-flow and congenital or infantile, including VOGM, AV fistulas, or rarely parenchymal bAVMs that are pathologically distinct from sporadic bAVMs. Therefore, precision medicine approaches must account for the distinct genetic drivers, mechanisms, and pathophysiological characteristics of syndromic bAVMs.
Human genetics of sporadic bAVMs. Pathogenic somatic mutations in KRAS, and less frequently BRAF, have been found in approximately 50%–80% of surgically resected, sporadic human bAVMs (8–10). A summary of human genetic studies on sporadic bAVMs is shown in Table 2. Nikolaev et al. first identified the role of somatic activating mutations in KRAS (G12D, G12V, and Q61H) (10). These mutations are found at low allele frequencies (0.5%–4%), are localized to ECs, and lead to MAPK/ERK hyperactivation. Reproduction of somatic KRAS mutations (G12D and G12V) and the discovery of new variants (G12A, G12C, and S65_A66insDS) were reported in separate human cohorts (8, 9, 85), in which G12A and G12C were also identified in patients with spinal AVMs (9).
Germline SNPs and de novo variant (DNV) analysis of human bAVMs reveal additional possible genetic contributions, but the causative evidence for these variants is lacking. For example, germline polymorphisms in angiopoietin-like 4 (ANGPTL4) (86) and DNVs across 46 genes converging on endoMT and on TGF-β/SMAD and VEGF signaling pathways have been implicated in bAVM risk (49, 87). Moreover, DNV analysis of 152 sporadic bAVM proband-parent trios and 40 singletons implicated multiple genes, including ANGPTL3 and SLC19A3 (88), whereas an independent cohort of 60 sporadic bAVM proband-parent trios identified nonsynonymous germline variants in 46 genes enriched in vascular cell types and involved in endoMT, including EXPH5, EPAS1, and ENG (87). Finally, in a series of 112 additional sporadic bAVM proband-parent trios, compound heterozygous variants in 16 genes were implicated in more than one trio, including five genes (MYLK, HSPG2, PEAK1, PIEZO, and PRUNE2) associated with angiogenesis or vascular disease (89). However, most of these variants have not been replicated or mechanistically validated (Table 2).


-
Single-cell analysis of the cerebrovasculature and bAVMs
Construction of large-scale cellular and transcriptomic atlases highlights the diversity, complexity, and heterogeneity of normal human CV system across development and disease (90–93). However, critical limitations to single-cell and spatial approaches in human bAVMs include surgical sampling bias to nidal tissue without parenchyma, reliable detection of low-frequency mutant ECs, lack of hemodynamic context, and a cross-sectional analysis without temporal developmental insights. Nonetheless, recent single-cell RNA sequencing (scRNA-seq) and supporting molecular validation experiments have identified changes within the CV in bAVMs, including accumulation of arterially fated and venous cluster ECs, presumably due to loss of capillary EC populations (a hallmark histopathological feature of bAVMs) (92). Mechanistically, Wälchli et al. demonstrated reactivation of a fetal gene signature, including multiple angiogenic pathways and angiogenic capillary markers PVLAP (and ESM1) that may be reflective of PVLAP’s role as an immature and non–BBB-competent EC marker (94). Moreover, these authors demonstrate disruption of the BBB and brain EC identities underscoring bAVM pathogenesis (92). Additional data in KrasG12D-driven bAVMs in mice also suggests that an increase in the tip cell population and angiogenic markers may be driven by androgen- and Notch-dependent signaling, providing rationale for investigation of therapeutic modulation of these pathways (95). An independent single-cell analysis of 106,853 cells across five resected bAVM specimens revealed 11 unique cell types, an increase in angiogenic potential and endoMT within the nidus, a unique immune cell signature including recruitment of myeloid cells after bAVM rupture, and modulation of hemorrhage risk via immune-mediated crosstalk with mural cells (93). Independent scRNA-seq in 17 resected bAVMs identified three unique EC clusters and one mural/fibroblast cluster (87). Finally, differential CpG methylation in EPHB1 and KRAS (among other genes) has been observed in human bAVM ECs (96). Taken together, these single-cell data frame bAVMs as lesions driven by EC identity, augmented AV developmental trajectories, BBB disruption, and pathologic inflammation coordinating EC–mural cell crosstalk contributing to rupture pathogenesis (Figure 2). Future efforts to resolve the mechanistic consequences of mosaic patterning of mutant cells and integration of bAVM genotype-phenotype molecular programs will inform therapeutic strategies.
 Figure 2
Figure 2Single-cell architecture of normal CV development and bAVM pathogenesis. (A) Normal CV development is characterized by carefully choreographed arterial-capillary-venous development through coordinated angiogenic sprouting and endothelial specificity. Mesenchymal and vascular smooth muscle cells (SMCs) provide structural support. (B) Pathogenic bAVM angiogenesis is characterized by loss of the intervening capillary bed and accumulation of arterial and venous ECs. Nidal tissue is characterized by high flow, immune cell recruitment, and expression of developmentally restricted angiogenic and BBB molecular programs. EC–mural cell crosstalk, unique inflammatory cell population recruitment, and endoMT characterize bAVM rupture states.

-
Models to elucidate bAVM pathogenesis and treatment paradigms
Many genetically engineered mouse and zebrafish models have been developed to elucidate syndromic (Table 3) and sporadic (Table 4) bAVM pathogenesis, each offering complementary strengths and limitations (97, 98). Syndromic (i.e., HHT and CM-AVM) models have been valuable in defining mechanisms underlying human disease genes, including TGF-β/BMP changes, and the interaction between inherited genetic factors and angiogenic stimuli. Consistent with the human disease, many syndromic models display systemic AVMs prone to rupture, confounding mortality and therapeutic success specific to bAVMs. In contrast, the zebrafish models depend on transient morpholinos and exhibit embryonic development of AVM-like lesions in embryonic vessel correlates that do not fully recapitulate the postnatal human disease (Table 3).
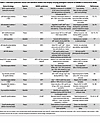 Table 3
Table 3Inherited syndromic mouse and zebrafish models that display varying pathogenic features of bAVMs or extracranial AVMs
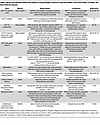 Table 4
Table 4Mouse and zebrafish models that display varying pathogenic features of sporadic bAVMs, extracranial AVMs, or bAVMs, and other AVM-like features
Since most human bAVMs are sporadic and driven by somatic KRAS — or less commonly BRAF — mutations, models expressing EC-specific human variants have provided critical mechanistic and therapeutic insights (Table 4). While no current model (adeno-associated virus–mediated [AAV-mediated] delivery or Cre-inducible transgenics) replicates the mosaicism and low pathogenic allele frequency seen in human sporadic bAVMs, they have been valuable in identifying a downstream dependency on MEK hyperactivation and for testing pharmacologic and gene editing strategies. However, these models likely overestimate disease penetrance, growth kinetics, and therapeutic response owing to the challenge of targeting rare cell populations, presence of approximately 1–3 bAVMs per mouse, and CV dysplasia beyond the bAVM in many cases. Nonetheless, EC-specific expression of KrasG12D or KrasG12V in mice results in multifocal extracranial and bAVMs (80, 99), and mice harboring brain-EC-specific expression of the driver mutation have been developed to overcome this limitation (100). Treatment of KrasG12D and KrasG12V mutant mice (Cre- or AAV-driven) with trametinib (MEK inhibitor) halted bAVM progression, reduced bAVM rupture, and normalized CV architecture (100–103). In addition, mice harboring EC-specific KrasG12C expression developed systemic and bAVMs that were ameliorated with sotorasib, a chemotherapeutic targeted therapy against the KrasG12C mutation (85). Overall, both syndromic and sporadic bAVM models provide a robust and reproducible means to define bAVM disease mechanisms and assess proof-of-principle therapeutic strategies.


-
Gene editing approaches for bAVMs
While molecularly targeted pharmacotherapies for bAVMs are appealing, gene editing technologies could, in principle, offer even greater precision, lasting effects, and a one-time genetic treatment (Figure 3). Limitations of CRISPR gene editing include editing specificity, targeting scope (determined by protospacer-adjacent motif [PAM] requirements), controlling and improving editing outcomes, and cell-targeted delivery methods (6, 7). Approaches to overcome specificity challenges include development of high-fidelity nuclease variants (114–116), modulating the duration of expression of the editor (117), and use of modified gRNAs (118, 119). The canonical 5′-NGG-3′ PAM sequence requirement of the Cas9 protein reduces targeting scope, but can be overcome using modified Cas9 or alternative Cas proteins with varying PAM requirements/specificities (120–130). Beyond traditional nuclease-based editing approaches, base editors can also introduce single nucleotide changes (131–134), and polymerization-based technologies, including prime editors (135) or DNA-dependent DNA polymerase–based editors (136, 137), can direct genomic DNA changes using RNA or DNA templates, respectively (Figure 3). These methods allow for more precise control of editing, particularly in the case of base or prime editing for single-point-mutation correction. Finally, additional gene editing tools include RNA editors (138) and epigenome editors (139, 140) that could be deployed in tandem with point-mutant-correction therapies (i.e., reduction of KRAS-dependent signaling) or as second-line therapies to modulate signaling pathways (i.e., VEGF, Notch, etc.) driving human bAVM maintenance, regression, response to radiation, and/or recurrence.
 Figure 3
Figure 3Gene editing strategies for bAVMs. (A) Strategies include CRISPR/Cas-based methods for gene knockout with nucleases to create insertions or deletions (indels) or templated edits via homology-directed repair. (B and C) Alternatively, more precise edits can be made with base editors and prime editors. (D) Delivery methods encompass lipid nanoparticles and (cerebro)vascular-specific AAVs, among others.
There are many potential challenges to gene editing for bAVMs. Specifically, human sporadic bAVMs display EC-restricted low variant allele fraction and mosaicism, requiring highly efficient and spatially restricted EC delivery. Recent advances in AAV engineering have expanded tropism to ECs in the cerebrovasculature (141–145), including a modified AAV-PR variant that transduces ECs and perivascular cells in murine models (144, 146). Lipid nanoparticles are an alternative approach, but important limitations include BBB permeability, where optimization of particle size, electrostatic properties, and selection of brain-EC-specific targeting ligands are potential clinical barriers (147–149). Nonetheless, in proof-of-concept experiments, CRISPR/Cas-mediated repression of KRAS expression has been shown to suppress bAVM growth and improve survival in KrasG12D mutant mice (99). Delivery method, immunogenicity, unintended nucleotide base conversions, introduction of genomic structural variations, and bystander edits stemming from nonspecific Cas9 binding or deaminase activity leading to unintended cellular perturbations remain key safety concerns for human clinical implementation. These data suggest that bAVMs may be suitable lesions for gene editing therapies, but a careful understanding of the disease biology, angioarchitectural markers of treatment success, and reliable detection and monitoring of the mutant EC fraction in situ will be necessary to guide the optimal gene editing approach, patient selection, and timing of treatment, among other factors.

-
Integrated clinical-genomic framework for targeted therapy in bAVMs
How would precision medicine approaches be implemented in human patients? Rational selection of targeted therapies for bAVMs (deemed inoperable, untreatable with SRS, or persistent despite SRS) requires a minimally invasive method (i.e., liquid and/or endoluminal biopsy) for detecting somatic pathogenic variants. We suggest a precision medicine approach for bAVMs that includes (a) patient-parent trio whole-exome sequencing to detect germline mutations, (b) in situ bAVM somatic genotyping, and to guide (c) eligibility for mutation-specific or pathway modulatory agents or (e) bespoke gene editing strategies to correct pathogenic somatic mutations (Figure 4). However, consensus is needed on defining clinical, angiographic, and possibly molecular endpoints, patient selection criteria, and comparisons with natural history studies.
 Figure 4
Figure 4Integrated clinical-genomic framework for precision medicine in bAVMs. Molecular diagnostics enabled by endoluminal and/or liquid biopsy for in situ detection of pathogenic somatic driver mutations to guide selection of molecularly targeted pharmacotherapy (sotorasib, vemurafenib, trametinib, etc.) or to identify pathogenic alleles for gene correction therapy in vivo (i.e., bespoke base editing approaches). Endoluminal and/or liquid biopsy can then be leveraged for postoperative bAVM surveillance in tandem with neuroimaging to monitor bAVM regression over time.

Article tools
- Download citation information
- Send a comment
- Terms of use
- Standard abbreviations
- Need help? Email the journal
Metrics
Go to
- Top
- Abstract
- Introduction
- Clinical overview of bAVMs
- Angiogenesis, AV specification, zonation, and CV development
- Genetic basis of bAVM pathogenesis
- Single-cell analysis of the cerebrovasculature and bAVMs
- Models to elucidate bAVM pathogenesis and treatment paradigms
- Molecularly targeted therapies for bAVMs
- Gene editing approaches for bAVMs
- In situ somatic mutation detection in CV pathology
- Integrated clinical-genomic framework for targeted therapy in bAVMs
- Conclusions
- Conflict of interest
- Funding support
- Acknowledgments
- Footnotes
- References
- Version history



Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)