Advertisement
Clinical Research and Public HealthAIDS/HIVCardiologyInflammation
Open Access | ![]() 10.1172/JCI196021
10.1172/JCI196021
Statin-dependent and -independent pathways are associated with major adverse cardiovascular events in people with HIV
Márton Kolossváry,1 Irini Sereti,2 Markella V. Zanni,1 Carl J. Fichtenbaum,3 Judith A. Aberg,4 Gerald S. Bloomfield,5 Carlos D. Malvestutto,6 Judith S. Currier,7 Sarah M. Chu,1 Marissa R. Diggs,1 Alex B. Lu,1 Christopher deFilippi,8 Borek Foldyna,9 Sara McCallum,1 Craig A. Sponseller,10 Michael T. Lu,9 Pamela S. Douglas,11 Heather J. Ribaudo,12 and Steven K. Grinspoon1
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Kolossváry, M. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by
Sereti, I.
in:
PubMed
|
Google Scholar
|

1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Zanni, M. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by
Fichtenbaum, C.
in:
PubMed
|
Google Scholar
|
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by
Aberg, J.
in:
PubMed
|
Google Scholar
|
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Bloomfield, G. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by
Malvestutto, C.
in:
PubMed
|
Google Scholar
|
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by
Currier, J.
in:
PubMed
|
Google Scholar
|
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Chu, S. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Diggs, M. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Lu, A. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by deFilippi, C. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Foldyna, B. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by McCallum, S. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Sponseller, C. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Lu, M. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by
Douglas, P.
in:
PubMed
|
Google Scholar
|
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Ribaudo, H. in: PubMed | Google Scholar
1Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
2Laboratory of Immunoregulation, National Institute of Allergy and Infectious Diseases (NIAID), NIH, Bethesda, Maryland, USA.
3Division of Infectious Diseases, University of Cincinnati College of Medicine, Cincinnati, Ohio, USA.
4Division of Infectious Diseases, Icahn School of Medicine at Mount Sinai, New York, New York, USA.
5Department of Medicine, Duke Global Health Institute and Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
6Division of Infectious Diseases, Ohio State University Medical Center, Columbus, Ohio, USA.
7Division of Infectious Diseases, David Geffen School of Medicine, UCLA, Los Angeles, California, USA.
8Inova Schar Heart and Vascular, Falls Church, Virginia, USA.
9Cardiovascular Imaging Research Center, Massachusetts General Hospital and Harvard Medical School, Boston, Massachusetts, USA.
10Kowa Pharmaceuticals America Inc., Montgomery, Alabama, USA.
11Duke Clinical Research Institute, Duke University School of Medicine, Durham, North Carolina, USA.
12Center for Biostatistics in AIDS Research, Harvard T.H. Chan School of Public Health, Boston, Massachusetts, USA.
Address correspondence to: Steven K. Grinspoon, Metabolism Unit, Massachusetts General Hospital and Harvard Medical School, 50 Staniford Street, 7th Floor, Office 768, Boston, Massachusetts 02114, USA. Phone: 617.724.9109; Email: sgrinspoon@mgh.harvard.edu.
Find articles by Grinspoon, S. in: PubMed | Google Scholar
Published September 9, 2025 - More info
J Clin Invest. 2025;135(22):e196021. https://doi.org/10.1172/JCI196021.
© 2025 Kolossváry et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: May 28, 2025; Accepted: August 28, 2025
-
Results
Study population. Between 2015 and 2018, a total of 804 participants were randomized in the REPRIEVE mechanistic substudy. For our current analysis, 39 individuals were excluded because of a lack of availability, sampling time, and/or quality of proteomics measurements, resulting in 765 individuals (379 randomized to pitavastatin and 386 to placebo, respectively) being included in our analyses (Figure 1). The average age of our participants was 50.8 ± 5.9 years, and 82% (n = 631) were men and 18% (n = 134) were women. The individuals were predominantly White, non-Hispanic participants or Latino participants. The mean duration of antiretroviral therapy (ART) was 11.8 ± 6.6 years at baseline. The average 10-year ASCVD risk score for the individuals was 5.0% ± 3.1%, and the baseline levels of LDL-C were 108.2 ± 29.8 mg/dL and 133.7 ± 83.2 mg/dL for triglycerides (Table 1).
 Figure 1
Figure 1CONSORT diagram of individuals included in the current analysis. Overall, 765 individuals were included in our analyses to evaluate the association between proteomics markers and MACE. QA, quality assurance; QC, quality control.
Over the average follow-up of 5.8 ± 1.9 years in the mechanistic substudy (median: 6.2 years, IQR: 5.5; 7.1 years), 33 participants experienced a MACE. Participants who experienced MACE were older (53.2 ± 5.4 vs. 50.7 ± 6.0 years, respectively), had higher 10-year ASCVD risk scores (6.9 ± 3.6 vs. 5.0 ± 3.1, respectively), were more likely to be a current smoker (52% vs. 24%, respectively), and had higher high-sensitivity C-reactive protein levels (4.5 ± 3.3 vs. 2.8 ± 2.9 mg/L, respectively, Table 1). In our analysis population, 20 individuals had a hard MACE (n = 6 cardiovascular deaths, n = 5 myocardial infarctions, n = 9 strokes, Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI196021DS1).
Association of statin-responsive proteins with MACE. Among a previously identified set of 7 proteins changing in response to pitavastatin treatment (6), 4 proteins showed decreased expression in response to statin therapy, and 3 showed increased expression. In our time-updated Cox models utilizing both baseline and 2-year proteomics measurements, only ANGPTL3 had a significant association with MACE (HR: 2.20 per 2-fold higher protein level; 95% CI: 1.04–4.67; P = 0.04, which corresponds to a HR of 1.40 and a 95% CI of 1.02–1.92 per population SD increase in normalized protein expression [NPX] values), such that lower levels of ANGPTL3 achieved with statin therapy were associated with lower MACE rates. Adjusting our models for 10-year ASCVD risk score and statin use resulted in similar results (HR: 2.31; 95% CI: 1.11–4.80; P = 0.03, which corresponds to a HR of 1.43 and a 95% CI of 1.04–1.96 per population SD increase in NPX values). No other proteins were significantly associated with MACE (Table 2). ANGPTL3 was also significantly associated with hard MACE (HR: 3.27; 95% CI: 1.32-8.13; P = 0.01, which corresponds to a HR of 1.66 and a 95% CI of 1.13–2.45 per population SD increase in NPX values), and this relationship remained consistent in adjusted models accounting for a 10-year ASCVD risk score and statin use (HR: 3.46; 95% CI: 1.43–8.53; P = 0.005, which corresponds to a HR of 1.70 and a 95% CI of 1.17–1.48 per population SD increase in NPX values). Supplemental analyses using statin randomization rather than time-updated statin use showed similar results (Supplemental Table 2).
In our study population, individuals randomized to statin therapy demonstrated an 11.1% (95% CI: –15.3%; –6.8%, P < 0.0001) reduction in ANGPTL3 levels, 15.3 mg/dL (95% CI: –27.4 mg/dL; –3.1 mg/dL; P = 0.013) reduction in triglyceride levels and a 27.8 mg/dL (95%CI: –32.5 mg/dL; –23.1 mg/dL; P < 0.001) reduction in LDL-C levels from baseline to 2-year follow-up compared with those randomized to placebo. In mediation analysis, 29.8% (95% CI: 11.6%; 52.5%) of the association between statin therapy and triglyceride levels and 5.9% (95% CI: 1.87%; 10.8%) of the association between statin therapy and LDL-C levels appeared mediated through the effect of statins on ANGPTL3. We thus further adjusted our analyses relating ANGPTL3 to MACE for time-updated LDL-C and triglyceride levels, which revealed similar results and a persistent relationship of ANGPTL3 to MACE (Supplemental Table 3).
Inflammatory, immune-oncological, and cardiometabolic proteomics markers related to MACE in residual risk analyses. Among the 248 remaining proteins in the proteomics panels (Supplemental Figure 1), which did not significantly change with statin treatment, in our time-updated Cox models utilizing both baseline and 2-year proteomics measurements, 26 proteins were associated with MACE at an FDR of less than 0.05 after adjusting for statin use and 10-year ASCVD risk scores. Among these, the receptor for IL-7 (IL-7R) (HR: 0.29; 95% CI: 0.13; 0.65; P = 0.003, which corresponds to a HR of 0.59; 95% CI: 0.42–0.83 per population SD increase in NPX values) and paraoxonase 3 (PON3) (HR: 0.45; 95% CI: 0.29; 0.68; P = 0.0001, which corresponds to a HR of 0.55; 95% CI: 0.41–0.75 per population SD increase in NPX values) were associated with a lower risk of MACE. All other proteins were associated with an increased the risk of MACE. The effect sizes of the remaining 24 proteins ranged from a HR of 1.58–9.20 per protein doubling (Figure 2 and Supplemental Table 4). We observed similar results in supplemental analyses adjusting for statin randomization rather than time-updated statin use (Supplemental Figure 2). Numerically, we also observed similar results for hard MACE (Supplemental Table 5). Among the 12 proteins with an FDR of less than 0.05 association with hard MACE, 10 were also identified for MACE. Only EGF-containing, fibulin-like extracellular matrix protein 1 (EFEMP1) and retinoic acid receptor responder protein 2 (RARRES2) were identified as associated with hard MACE but did not reach statistical significance for the MACE outcome. Detailed results, protein names, and abbreviations for proteins identified in these analyses can be found in Table 3. In sensitivity analyses, further adjustment for LDL-C and triglyceride levels produced numerically similar results for all proteins (Supplemental Figure 3).
 Figure 2
Figure 2Association between proteomics markers and MACE or hard MACE. (A) Volcano plot of HRs and P values for given proteins and MACE. HRs were estimated from Cox proportional hazards models using both potential proteomics measurements including statin use as a time-dependent covariate and 10-year baseline ASCVD risk score. HRs are per protein doubling. Red proteins indicate associations at an FDR below 0.05 level. The x axis is on a log2 scale. Dotted line indicates a nominal P value of 0.05. Dashed line indicates a HR of 1. (B) Volcano plot of HRs and P values for given proteins and hard MACE. Hard MACE is defined as cardiovascular death, myocardial infarction, or stroke. HRs were estimated from Cox proportional hazards models using both potential proteomics measurements including statin use as a time-dependent covariate and baseline 10-year ASCVD risk score. HRs are per protein doubling. Red proteins indicate associations at an FDR below 0.05. The x axis is on a log2 scale. Dotted line indicates a nominal P value of 0.05. Dashed line indicates a HR of 1. ANGPT2, angiopoietin-2; CD70, CD70 antigen; CD83, CD83 antigen; CSF1, processed macrophage colony-stimulating factor 1; CXCL13, C-X-C motif chemokine 13; FABP4, fatty acid–binding protein, adipocyte; GAL9, galectin-9; GRN, paragranulin; HGF, hepatocyte growth factor α chain; MCP2, C-C motif chemokine 8; MCP3, C-C motif chemokine 7; NOS3, nitric oxide synthase, endothelial; NT-PROBNP, NT-proBNP; OPN, osteopontin; PDCD1, programmed cell death protein 1; SHPS1, tyrosine-protein phosphatase non-receptor-type substrate 1; TCN2, transcobalamin-2; TNFRSF9, TNF receptor superfamily member 9; TNFSF13B, TNF ligand superfamily member 13b, membrane form; VEGFA, vascular endothelial growth factor A.
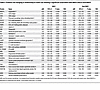 Table 3
Table 3Proteins not changing in relationship to statin use showing a significant association with MACE and/or hard MACE
Correlation among proteins related to MACE and relation to baseline clinical factors. ANGPTL3 was related to multiple proteins and, most significantly, to the urokinase plasminogen activator surface receptor (UPAR) (ρ = 0.54). In contrast, other proteins related to MACE showed only a moderate correlation with each other. Proteins related to MACE were only weakly correlated with clinical factors and clinically assessed biomarkers. A detailed correlation heatmap can be found in Supplemental Figures 4 and 5.
Protein-protein interaction analysis for non-statin-modifiable proteins. For the 28 proteins showing a significant association with MACE or hard MACE at an FDR below 0.05 in residual risk analyses, we created an exploratory protein-protein interaction network to further understand the biological role of these proteins. All proteins except PON3 and transcobalamin-2 (TCN) were part of a single cluster (average number of connections: 6.21), where IL-6 and IL-8 were connected to the largest numbers of other proteins (n = 21 and n = 18, respectively, Figure 3). Exploratory Gene Ontology biological processes enrichment analysis showed an increased representation of leukocyte-associated chemotaxis and migration and humoral immune response. Gene Ontology molecular function enrichment analysis indicated increased cytokine- and chemokine-associated functions in our network. Reactome pathway analysis indicated increased enrichment of TNF, cytokine, and chemokine pathway functions within our significant proteins. Detailed enrichment results can be found in Figure 3, with the lists of proteins corresponding to each term in Supplemental Table 6.
 Figure 3
Figure 3Protein-protein interaction and enrichment analysis among proteins showing a significant association with MACE or hard MACE. (A) Confidence-based protein interaction network using all available interactions sources. The width of the edges between the proteins represents the overall confidence score. The average number of connections between the protein nodes is 6.21, while IL-6 and IL-8 are connected to 21 and 18 proteins, respectively. (B) Gene Ontology and Reactome functional enrichments analysis. Gene Ontology biological processes, molecular function, and Reactome pathway enrichment all indicated increased representation of cytokine and chemokine processes and pathway functions within our significant proteins. CCR, chemokine-chemokine receptor.
Diagnostic power of proteomics markers to predict MACE beyond traditional risk indices. To account for collinearity between the proteomics features, avoid overfitting, and evaluate the additive value of significant proteomics features, we built a machine learning elastic net Cox proportional hazards model using the significant proteomics markers, treatment randomization, and 10-year ASCVD risk scores, applying stratified 10-fold cross-validation. We developed 3 models. Model 1, incorporating statin randomization and the 10-year ASCVD risk score, achieved a 10-fold cross-validated concordance index (C-index) of 0.61 (SD = 0.18) for MACE. In contrast, model 2, which incorporated only baseline protein values, demonstrated a cross-validated C-index of 0.74 (SD = 0.12). Model 3, including ASCVD risk, statin randomization, and protein values, similarly demonstrated a cross-validated C-index of 0.74 (SD = 0.11). Lysosome-associated membrane glycoprotein 3 (LAMP3) had the largest HR among proteins positively associated with MACE, and PON3 had the lowest HR among proteins negatively associated with MACE (Figure 4).
 Figure 4
Figure 4Cross-validated performance of significant proteins, unchanged with statin therapy, to predict MACE. (A) Stratified 10-fold cross-validated, time-dependent C-index of Cox elastic net machine learning models. Model 1: statin randomization + 10-year ASCVD risk score; model 2: baseline values of proteins associated with MACE; model 3: model 1 + model 2. (B) Importance of the features with a HR different from at least 1 in one of the models. Bars represent standardized average standardized HRs from the 10-fold cross-validation. CV, cross-validation.










Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)