Advertisement
Research ArticleImmunologyPulmonology
Open Access | ![]() 10.1172/JCI188891
10.1172/JCI188891
Impaired complement regulation drives chronic lung allograft dysfunction after lung transplantation
Hrishikesh S. Kulkarni,1,2 Laneshia K. Tague,1 Daniel R. Calabrese,3,4 Fuyi Liao,5 Zhiyi Liu,5 Lorena Garnica,1 Nishanth R. Shankar,1 Xiaobo Wu,1 Devesha H. Kulkarni,2 Aayusha Thapa,2 Dequan Zhou,5 Yan Tao,1 Victoria E. Davis,5 Cory T. Bernardt,6 Derek E. Byers,1 Catherine Chen,7 Howard J. Huang,8 Chad A. Witt,1 Ramsey R. Hachem,9 Daniel Kreisel,5,6 John P. Atkinson,1 John R. Greenland,3,4 and Andrew E. Gelman5,6
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by
Kulkarni, H.
in:
PubMed
|
Google Scholar
|

1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by
Tague, L.
in:
PubMed
|
Google Scholar
|
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by
Calabrese, D.
in:
PubMed
|
Google Scholar
|
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Liao, F. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Liu, Z. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Garnica, L. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Shankar, N. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Wu, X. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Kulkarni, D. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Thapa, A. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Zhou, D. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Tao, Y. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Davis, V. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Bernardt, C. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by
Byers, D.
in:
PubMed
|
Google Scholar
|
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Chen, C. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by
Huang, H.
in:
PubMed
|
Google Scholar
|
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Witt, C. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Hachem, R. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Kreisel, D. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by Atkinson, J. in: PubMed | Google Scholar
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by
Greenland, J.
in:
PubMed
|
Google Scholar
|
1Department of Medicine, Washington University School of Medicine, St. Louis, Missouri, USA.
2Department of Medicine, David Geffen School of Medicine at UCLA, Los Angeles, CA, USA.
3Department of Medicine, UCSF, San Francisco, California, USA.
4Medical Service, Veterans Affairs Health Care System, San Francisco, California, USA.
5Department of Surgery and
6Department of Pathology & Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
7Department of Internal Medicine, University of Texas Southwestern Medical Center, Dallas, Texas, USA.
8Department of Medicine, Houston Methodist Hospital, Houston, Texas, USA.
9Department of Internal Medicine, University of Utah Spencer Fox Eccles School of Medicine, Salt Lake City, Utah, USA.
Address correspondence to: Hrishikesh Kulkarni, Division of Pulmonary, Critical Care and Sleep Medicine, University of California, Los Angeles, 10833 Le Conte Ave, 52-257 CHS, Mail code: 169017, Los Angeles, California 90095, USA. Email: hskulkarni@mednet.ucla.edu. Or to: Andrew E. Gelman, Mary Culver Department of Surgery, Division of Cardiothoracic Surgery, Washington University School of Medicine, 660 S Euclid Ave, St. Louis, Missouri 63110, USA. Phone: 314.362.8382; Email: agelman@wustl.edu.
Find articles by
Gelman, A.
in:
PubMed
|
Google Scholar
|
Published November 11, 2025 - More info
J Clin Invest. 2026;136(1):e188891. https://doi.org/10.1172/JCI188891.
© 2025 Kulkarni et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: November 18, 2024; Accepted: November 6, 2025

-
Results
A functional polymorphism in the complement component C3 gene is observed in at least 1 in 3 LTx recipients. Within the LTx cohort at Washington University/Barnes-Jewish Hospital (BJH), we identified 392 participants with available genotyping (Figure 1A). Among these recipients, 244 (62.2%) had wild-type C3 (G/G); thus, the recipient rs2230199 (G>C) SNP had a minor allele frequency (allele C) of 37.8%. Baseline characteristics for participants stratified by genotype are shown in Table 1 and were similar across both genotypes.
 Figure 1
Figure 1A functional C3 polymorphism confers increased risk of CLAD or death in 2 independent cohorts. Consolidated Standards of Reporting Trials diagrams for the Washington University/BJH (A) and UCSF cohorts (B). (C and E) Kaplan-Meier plot of survival from CLAD or death in the BJH (C) and UCSF (E) cohorts stratified by rs2230199 genotypes. (D and F) Kaplan-Meier plot based on the multivariate Cox regression analysis of CLAD-free survival in the BJH (D) and UCSF (F) cohorts by rs2230199 status. The P value was determined by log-rank test.
Within the UCSF LTx cohort, we identified 425 participants with available genotyping (Figure 1B). The recipient rs2230199 (G>C) SNP had a minor allele frequency (allele C) of 29.4%. Baseline characteristics for participants stratified by genotype are shown in Table 2. Notably, we found higher minor allele frequencies among White recipients and lower minor allele frequencies among Black recipients. These distributions are consistent with those reported in aggregated global genomic databases (40). There were no other differences in LTx baseline characteristics across genotypes.
C3 R102G confers increased risk of CLAD or death in 2 independent cohorts. We next examined whether the rs2230199 C3 minor allele, which predisposes to increased complement activation, was associated with decreased CLAD-free survival. In the BJH cohort, the C3 R102G polymorphism was associated with an increased risk for CLAD or death (Figure 1C). In a multivariable Cox-proportional hazards model consisting of C3 rs2230199 genotype, age, sex, race, transplant diagnosis, DSAs, and Gram-negative infection, C3 R102G remained associated with increased risk of CLAD or death (Figure 1D and Table 3). The presence of post-transplant DSAs (adjusted hazard ratio [aHR] 1.74; 95% CI, 1.24–2.45; P = 0.001) or post-transplant Gram-negative infection (aHR 1.44; 95% CI, 1.01–2.06; P = 0.044) also was associated with a significantly increased risk of CLAD or death. A sensitivity analysis removing the presence of DSAs among the list of variables demonstrated that C3 R102G was still associated with an increased risk of CLAD or death (aHR 1.52; 95% CI, 1.08–2.15; P = 0.016). A similar analysis removing the presence of Gram-negative infection among the list of variables demonstrated that C3 R102G remained associated with an increased risk of CLAD or death (aHR 1.43; 95% CI, 1.02–2.02; P = 0.04).
We examined if R102G also was associated with differential CLAD-free survival in the UCSF cohort. Figure 1E shows that participants homozygous for C3 R102G had an increased risk for CLAD or death (HR 1.43; 95% CI, 1–2.05; adjusted P = 0.048). This multivariable Cox-proportional hazards model also consisted of C3 rs2230199 genotype, age, sex, race, transplant diagnosis, DSAs, and Gram-negative infection (Figure 1F and Table 4).
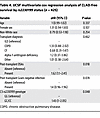 Table 4
Table 4UCSF multivariate cox regression analysis of CLAD-free survival by rs2230199 status (n = 425)
CLAD-free survival association with recipient C3 R102G is linked to DSAs. Based on work published by us and others reporting that anti-HLA DSAs are a known risk factor for CLAD (11, 41), we hypothesized that the increased risk of CLAD or death in recipients with C3 R102G would be limited to those with DSAs. In the BJH cohort, 181 LTx recipients (46.2%) had definite DSAs during their follow-up. CLAD-free survival analysis accounting for both DSAs and rs2230199 genotype demonstrated that recipients with C3 R102G and DSAs had the worst CLAD-free survival (Figure 2A). Among recipients without DSAs, we did not observe a difference in risk for CLAD or death based on the C3 rs2230199 genotype (HR 1.21; 95% CI, 0.72–2.04; P = 0.48). However, among the participants who developed DSAs and had the C3 R102G polymorphism, there was an increased risk for CLAD or death (HR 1.52; 95% CI, 0.99–2.34; P = 0.05), with the difference in CLAD-free survival primarily being observed starting at approximately 2.5 years after transplantation.
 Figure 2
Figure 2Complement-mediated CLAD or death is dependent on DSAs. CLAD-free survival was worse in the recipients with DSAs and the C3 R102G polymorphism (CC/GC). The BJH (A) and UCSF (B) cohorts were stratified by DSA-negative and DSA-positive recipients. The P value was determined by log-rank test. (C and D) Kaplan-Meier plot based on the multivariate Cox regression analysis of CLAD-free survival in the UCSF cohort (C and D) and BJH (E and F), separated by genotype and stratified by DSA status.
These observations also held true in the UCSF cohort, wherein 87 LTx recipients (20.5%) had evidence of DSAs at some point during their post-transplant course. Among participants without DSAs there was no different risk for CLAD or death by the rs2230199 genotype (aHR 1.2; 95% CI, 0.85–1.9; adjusted P = 0.24) (Figure 2B). However, among the participants who developed DSAs and had a CC or a GC allele, there was a 3.2-fold increased risk for CLAD or death (aHR 3.2; 95% CI, 1.6–6.2; adjusted P = 0.0007). These models were adjusted for age, sex, race, transplant diagnosis, and Gram-negative infection. These findings suggest the impact of the rs2230199 SNP is dependent upon DSA development.
We next examined CLAD-free survival separately in recipients based on the presence or absence of C3 R102G. Recipients in the UCSF cohort who lacked C3 R102G (i.e., GG genotype) did not demonstrate a significant difference in CLAD-free survival irrespective of their DSA status (Figure 2C). In comparison, recipients with post-transplant DSAs and the C risk allele (i.e., CG or CC genotype) were at a 3-fold higher risk for worse CLAD-free survival after adjusting for age, sex, race, transplant diagnosis, and Gram-negative infection (Figure 2D). The results also held true in the BJH cohort, in which recipients with post-transplant DSAs and the C risk allele had worse CLAD-free survival (aHR 1.9; 95% CI, 1.1–3.3; adjusted P = 0.02; Figure 2, E and F). These data suggest the worst long-term outcomes occurred in those who developed post-transplant DSAs and harbored the rs2230199 SNP.
Impaired complement regulation promotes CLAD in the mouse orthotopic LTx model. The C3 R102G polymorphism lacks a synonymous variant in mice. However, mice deficient in Crry (Crry–/–) have increased complement activation due to impaired Factor I–mediated cleavage of C3b and C4b, resulting in dysregulated C3 convertase formation on the membrane (38). As a result, these mice have more C3 consumption and lower circulating levels of C3 (39, 42), as well as attenuated levels of C3 in their bronchoalveolar lavage (BAL) (Figure 3A). Given our prior findings that C3 is required for epithelial cell survival (27) and that C3 R102G predisposes to impaired complement regulation (36), we asked if CLAD development is regulated by complement activation. For this purpose, we used a previously established orthotopic mouse LTx model of CLAD (43, 44) (Figure 3B). Wild-type Crry+/+ and Crry–/– recipients, both on a B6 background (H-2b), were engrafted with major-mismatched left lungs encoding 3 transgenes (3T-FVB; H-2q): a reverse tetracycline activator gene driven by the club cell secretory protein promoter, a Cre recombinase gene under the control of the reverse tetracycline activator, and a lox-P activated diphtheria toxin A gene. These transgenes induce graft-specific club cell injury and depletion in B6 recipients following 2.5 days of doxycycline (DOX) ingestion, leading to severe CLAD development with a combination of obliterative and restrictive fibrotic pathology that is dependent on alloimmune responses to a major histocompatibility class mismatch. In contrast, 30 hours of DOX ingestion only induces minimal club cell depletion and results in markedly less allograft injury.
 Figure 3
Figure 3Dysregulated complement activation promotes CLAD in a mouse LTx model. (A) Immunoblot of serum and BAL C3-α and -β fragments from resting Crry+/+ and Crry–/– mice (n = 4/group). The data shown are representative results of 2 experiments. (B) Mouse orthotopic left LTx model of CLAD. (C) H&E and Masson’s trichrome (MT) staining of a heart-lung block from POD 16 Crry+/+ and Crry–/– allograft recipients. Scale bars: 250 μm. The histology shown is a representative result of at least 6 transplants per group. (D) POD 16 blinded A (vascular) and B (airway) ISHLT consensus pathological scoring (n = 7/group). (E) immunofluorescent staining of POD 16 allograft club and ciliated cells using Abs specific for CCSP and acetylated tubulin (Ac-Tubulin). The images shown are representative results of 5 transplants per group. (F) A representative flow cytometry plot gating strategy for identifying and (G) quantifying POD 16 CD326+ CCSP+ (club) cells in POD 16 allografts (n = 4/group). (H) POD 16 intragraft total and indicated subset CD4+ and CD8+ T cell numbers (n = 4/group). Bar graphs and dot plots show mean ± SD for (D) Mann-Whitney U test and (G and H) Welch’s t test. *P < 0.05, ***P < 0.001. NL, native lung.
After induction of immunosuppression-mediated lung allograft acceptance, recipients ingested DOX on postoperative day (POD) 7 for 30 hours; on POD 16, allografts were examined for histological evidence of CLAD. We observed more evidence of peribronchiolar fibrosis, obliterative airway disease, scarring of the alveolar parenchyma, and collagen deposition in allografts of Crry–/– when compared with wild-type recipients (Figure 3C and Supplemental Figure 1). Blinded grading for LTx rejection using consensus International Society for Heart and Lung Transplantation (ISHLT) criteria (45) showed significantly higher “B” airway inflammation in allografts after transplantation into Crry–/– relative to wild-type recipients (Figure 3D). Consistent with these findings, club cells failed to fully reconstitute the graft epithelium in Crry–/– hosts. Some club cells were observed scattered throughout the interstitium, suggesting epithelial repair is inhibited by augmented complement activation (Figure 3, E–G). Similar to previous findings in humans with CLAD (46, 47), there were more intragraft Th17 and IFN-γ+ CD8+ T cells in mouse recipients lacking Crry (Figure 3H).
Impaired complement regulation promotes CLAD in a B cell–dependent manner. Previous work has shown that activated C3 fragments bind to B cells to enhance their activation (48, 49). Given these observations, we asked whether Crry deficiency was associated with increased B cell activity in our CLAD model. C3d deposition on B cells was increased in the spleen and allografts of Crry–/– recipients (Figure 4A). There were also more total and proliferating CD19+B220+ B cells in the allografts of Crry–/– recipients (Figure 4B). We also quantified B cell levels in human LTx recipients carrying the C3 R102G variant (Figure 4C). A subcohort of 218 recipients with C3 R102G had increased CD19+ B cell frequencies in their BAL fluid after adjusting for age, sex, diagnostic group, and ethnicity. Likewise, we observed higher numbers of B cells in the BAL fluid of Crry–/– compared with wild-type recipients (Figure 4D and Supplemental Figure 2). De-novo DSAs are associated with an increased risk for CLAD development and progression (50). Therefore, we next measured DSA titers in Crry–/– recipients. Relative to wild-type recipients, Crry–/– recipients had higher IgM, IgG2c, and IgG3 DSAs in their BAL fluid (Figure 4E). By contrast, only IgM DSA numbers were elevated in the peripheral circulation of Crry–/– recipients (Figure 4F). Because DSA accumulation in BAL fluid indicated the local production of Abs, we assessed the abundance of Ab-secreting cells within allograft tissue. Analysis of allografts of Crry–/– recipients revealed more intragraft IgM+ and IgG+ Ab-secreting cells relative to wild-type recipients (Figure 4G and Supplemental Figure 3). CD73, CD86, and CD273 co-expression defines a memory B cell subset poised to become Ab-secreting cells (51). Strikingly, only Crry–/– recipient allografts had IgM+ and IgG+ CD73+CD80+CD273+ memory phenotype B cells (Figure 4, H and I).
 Figure 4
Figure 4Enhanced intragraft B cell accumulation and activation in lung recipients with a defect in complement regulation. (A) Levels of C3d bound to B cells 2 days after the induction of DOX-induced allograft club cell injury. Representative FACS histogram of C3d B cell staining and (right panel) plots of C3d MFI normalized to a corresponding fluorescence minus one (FMO) control (n = 4/group). (B) POD 16 FACS contour plot of intragraft B cell abundance (left), ki67 histogram (proliferation) (middle), and a plot of total allograft B cell numbers on POD 16 (right). FACS contour plots and histograms are representative results from 4/group. (C) Percentages of BAL CD19+ lymphocytes in UCSF cohorts who carry the GG genotype and C3 R102G (CC&GC) polymorphisms. P = 0.02 by an unpaired t test. (D) Quantitation of BAL B cell numbers in POD 16 allografts (n = 4/group). Levels of POD 16 of (E) BAL (n = 4/group) and (F) serum (n = 5/group) donor reactive IgM, IgG1, IgG2c, and IgG3. (G) POD 16, intragraft CD138+ IgM+ and IgG+ Ab-secreting cell numbers (n = 5/group). (H) A representative FACS gating strategy used to identify intragraft CD80+CD73+CD273+ memory B cells, as shown in the bar graphs (I), which display the total intragraft numbers of these cells expressing either surface IgM (n = 5/group) or IgG (n = 4/group) on POD 16. Bar graphs and dot blots show mean ± SD values according to 2-way ANOVA with Šidák’s multiple comparisons test (A), and Welch’s t test (B, D–G, and I). *P < 0.05, **P < 0.01, ***P < 0.001.
Given that our data indicated dysregulated complement regulation triggers the production of DSAs, we next sought evidence of intragraft classical pathway activation by assessing C3d deposition. Relative to wild-type lung recipients, C3d deposition was highly prevalent on B cells within tertiary lymphoid aggregates and, to a lesser degree, on the bronchial epithelium and vascular endothelium in allografts of Crry–/– recipients (Figure 5, A and B). We also analyzed Ab deposition in the lung parenchyma. IgG deposition was more apparent on the allograft alveolar capillaries and bronchial epithelium of Crry–/– recipients relative to the native lungs of Crry–/– recipients or wild-type recipient allografts (Figure 6, A and B). Although IgM expression was present on cells within the pleural cavity, IgM deposition was almost undetectable on lung parenchyma (Supplemental Figure 4).
 Figure 5
Figure 5Lung allografts from Crry-deficient recipients show elevated C3d deposition. C3d and CD19 immunofluorescent staining of Crry–/– (A) and wild-type Crry+/+ (B) recipient allograft tissue (LTx). Stars and arrows denote C3d staining on vascular endothelium and bronchial epithelium, respectively. Rectangular insets depict tertiary lymphoid aggregates enriched with B cells. Scale bars: 150 μm. The images shown are representative results of at least 3 transplants per group.
 Figure 6
Figure 6IgG deposition is more pronounced on lung allograft parenchyma from Crry-deficient recipients. (A) Immunofluorescence detection of IgG and IgM deposition on alveolar capillaries, as denoted by CD31 Ab staining. Inset scale bars: 25 μm. White arrowheads denote IgG deposition on alveolar capillaries. Images shown are representative of 4 transplants per group. (B) IgG and IgM deposition analysis on allograft bronchioles (b) and vessels (v). Inset scale bars: 150 μm. Images shown are representative of 4 transplants per group.
Finally, to examine if B cells are required for CLAD development in our model, Crry–/– recipients were treated with B cell–depleting Abs and evaluated on POD 16. Compared with recipients that received control Abs, B cell depletion significantly inhibited pathological signs of CLAD development (Figure 7, A and B, and Supplemental Figure 5).
 Figure 7
Figure 7CLAD development in Crry-deficient allograft recipients is driven by B cells. (A) Crry-deficient lung recipients received B cell–depleting Abs or control Ig on PODs 6 and 12 and were evaluated for allograft injury on POD 16. Inset scale bars: 200 μm. The histology shown is a representative result of H&E and Masson’s trichrome (MT) allograft staining of 5 transplants per group. (B) POD 16 blinded A (vascular) and B (airway) ISHLT consensus pathological scoring (n = 5/group). The bar graph shows mean ± SD; the Mann-Whitney U test was used to generate P values.














Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)