Advertisement
Research ArticleInflammationNeuroscience
Open Access | ![]() 10.1172/JCI183393
10.1172/JCI183393
Gestational hypertension increases risk of seizures in children and mice
Baojian Xue,1 Serena B. Gumusoglu,2,3,4 Grant Tiarks,1 Brittany P. Todd,1 Angela Wong,1 Donna A. Santillan,2,4 Chin-Chi Kuo,5,6,7 Hsiu-Yin Chiang,5,6 Rohith Ravindranath,8 Sophia Y. Wang,8 Vinit B. Mahajan,9 Alan Kim Johnson,10 Heath A. Davis,11 Polly Ferguson,1 Elizabeth A. Newell,1 Mark K. Santillan,2,4 Jason M. Misurac,1 and Alexander G. Bassuk1,12,13
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Xue, B. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Gumusoglu, S. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Tiarks, G. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Todd, B. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Wong, A. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by
Santillan, D.
in:
PubMed
|
Google Scholar
|

1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Kuo, C. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Chiang, H. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Ravindranath, R. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Wang, S. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by
Mahajan, V.
in:
PubMed
|
Google Scholar
|
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Johnson, A. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Davis, H. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by
Ferguson, P.
in:
PubMed
|
Google Scholar
|
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Newell, E. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by
Santillan, M.
in:
PubMed
|
Google Scholar
|
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by Misurac, J. in: PubMed | Google Scholar
1Stead Family Department of Pediatrics,
2Department of Obstetrics and Gynecology,
3Department of Psychiatry, and
4The Hawk Intellectual and Developmental Disabilities Research Center, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
5Big Data Center,
6Department of Biomedical Informatics, and
7Department of Internal Medicine, China Medical University Hospital, Taichung, Taiwan.
8Byers Eye Institute, and
9Molecular Surgery Laboratory, Stanford University, Palo Alto, California, USA.
10Department of Psychological and Brain Sciences,
11Institute for Clinical and Translational Science-Biomedical Informatics,
12The Iowa Neuroscience Institute, and
13The Department of Neurology, Roy J. and Lucille A. Carver College of Medicine, University of Iowa, Iowa City, Iowa, USA.
Address correspondence to: Alexander G. Bassuk, Stead Family Department of Pediatrics, The University of Iowa, 25 S. Grand Ave., 2040 Medical Laboratories, Iowa City, Iowa, 52242, USA. Email: alexander-bassuk@uiowa.edu.
Find articles by
Bassuk, A.
in:
PubMed
|
Google Scholar
|
Published June 16, 2025 - More info
J Clin Invest. 2025;135(12):e183393. https://doi.org/10.1172/JCI183393.
© 2025 Xue et al. This work is licensed under the Creative Commons Attribution 4.0 International License. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Received: May 29, 2024; Accepted: April 9, 2025

-
Results
Increased seizures among children from hypertensive pregnancies in 4 large clinical datasets. To first identify whether there is an overall association between maternal hypertension exposure and childhood seizure, we used the Epic Cosmos database of 246 million individuals treated across 1,400 hospitals and 33,000 clinics in the United States and Lebanon. We identified 229,357 individuals (below 18 years of age) with a diagnosis of seizures among 7,257,078 children with birth parent data available. The rate of seizure was 50,288 of 1,365,254 (3.68%) in patients born to mothers who were hypertensive during pregnancy and 179,069 of 5,891,824 (3.04%) in patients born to mothers without hypertension during pregnancy. The χ2 contingency test indicated that exposure to maternal hypertension was significantly associated with a higher incidence of seizures (OR 1.22; 95% CI: 1.21–1.23, P < 0.001) in children. Moreover, male offspring were overrepresented relative to female offspring among the seizure groups (P < 0.001), regardless of maternal hypertension.
While these analyses did not produce line-level data allowing for covariate adjustment, we found that the rate of obesity was higher in mothers with hypertension than in those without (43.0% vs 16.3%; P < 0.001), as was maternal diabetes (20.4% vs 8.4%; P < 0.001). We also found that the rate of developmental delay was higher in children from a hypertensive pregnancy than in those from normotensive pregnancies (10.3% vs. 7.7%; P < 0.001), an association observed in children either with (31.3% vs 25.8%; P < 0.001) or without (9.5% vs 7.1%; P < 0.001) a seizure diagnosis. Full demographic information and statistical comparisons are presented in Table 1 and Supplemental Tables 1–4 (supplemental material available online with this article; https://doi.org/10.1172/JCI183393DS1).
 Table 1
Table 1Cosmos population characteristics and ORs show that maternal hypertension during pregnancy is a risk factor for seizure in children
To determine whether the association between GH and child seizure risk survived covariate adjustment, we next examined this association within a richly annotated clinical knowledgebase — the IGHK. In this database, pregnant women whose neonates experienced seizures (n = 1,370) had significantly higher rates of maternal body mass index (BMI) (16% vs. 12%; P < 0.001), adverse neonatal outcomes (59% vs. 35%; P < 0.001), and HDP (37% vs. 33%; P < 0.001) compared with controls (n = 34,297). After controlling for age, gravida, BMI, diabetes, and ancestry, pregnant women affected by HDP had higher odds of having a child with seizures than did pregnant women without HDP (adjusted OR = 1.132 [1.003–1.278], P < 0.05) (Table 2 and Supplemental Tables 5 and 6).
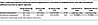 Table 2
Table 2IGHK study regression shows that maternal hypertension is a risk factor for childhood seizure, even after controlling for additional covariates by logistic regression
We next expanded our study to another large, US-based clinical data repository: the Stanford Research Data Repository Electronic Health Records Observational Medical Outcomes Partnership (STARR-OMOP). The STARR-OMOP is the Stanford Electronic Health Records research database consisting of linked maternal-child data from both the adult Stanford Healthcare system and the Lucile Packard Children’s Hospital system. In this dataset, maternal hypertension significantly predicted a child’s (<18 years of age) risk of seizures, even after adjusting for maternal age, maternal BMI above 40, diabetes in pregnancy, and ancestry (adjusted OR 1.36; 95% CI: 1.21–1.52, P < 0.001). Full cohort characteristics and statistics are presented in Table 3 and Supplemental Tables 7 and 8.
 Table 3
Table 3Stanford validation study regression shows that maternal hypertension is a risk factor for childhood seizure, even after controlling for additional covariates by logistic regression
Finally, to test whether maternal hypertension is associated with child seizures across diverse settings, we examined this association in a large international cohort: Taiwan’s National Health Insurance Database of 2,003,354 mother-child pairs. After adjusting for maternal age, maternal diabetes, maternal obesity, gestational age at delivery, infant birth weight, infant 5-minute Apgar score, and childhood development delay, we found that maternal hypertension was significantly associated with an increased risk of childhood seizure (adjusted OR 1.17; 95% CI: 1.14–1.20, P < 0.001). Full cohort characteristics and statistics are presented in Table 4 and Supplemental Tables 9–11.
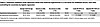 Table 4
Table 4Taiwanese validation study regression shows that maternal hypertension is a risk factor for childhood seizure, even after controlling for covariates by logistic regression
Together, our studies across 4 complementary cohorts (Epic Cosmos, IGHK, Stanford STARR-OMOP, and Taiwan’s National Health Insurance Database) link maternal hypertension exposure to pediatric seizure occurrence in the next generation.
ANG II or PE is sufficient to cause hypertension in murine pregnancy. To complement our clinical analyses, we next utilized 2 complementary animal GH models, the chronic ANG II model and the PE model. Both models have been validated to induce hypertension in mice. There were no differences in baseline blood pressure (BP) or heart rate (HR) between saline- and ANG II–treated dams. During pregnancy, systolic BP, diastolic BP, and mean arterial pressure (MAP) were significantly increased in ANG II-infused females compared with saline-infused control females (P < 0.05, Figure 1A and Supplemental Figure 1), but there were no differences in HR or locomotor activity (Figure 1B and Supplemental Figure 1). ANG II–induced GH did not impair reproduction or skew offspring sex ratios. In total, 31 dams with ANG II–induced hypertension produced 200 pups including 102 males and 98 females, whereas 39 normotensive dams produced a total of 263 pups including 134 males and 129 females. There were no significant differences in litter size by condition (6.7 ± 0.3 pups vs. 6.5 ± 0.2 pups, Figure 1C). However, the average weight of ANG II pups on the day of birth (P0) was less than that of control pups (1.33 ± 0.03 g/pup vs. 1.43 ± 0.02 g/pup, respectively, P < 0.05, Figure 1D).
 Figure 1
Figure 1Maternal ANG II– or PE-induced GH and their effects on the neonates. Changes in MAP (A) and HR (B) in mouse dams with chronic infusion of saline, ANG II, or PE during pregnancy. (C and D) Litter size and birth weights. Two-way ANOVA; *P < 0.05 vs. saline dams; #P < 0.05 vs. PE-treated dams; †P < 0.05 vs. pups of saline-treated dams. Data are shown as the mean ± SEM.
In the PE-induced GH model, 10 dams with PE-induced hypertension produced 58 pups including 31 males and 27 females. PE infusion in pregnancy significantly increased MAP and systolic BP, though not to the degree of that induced by ANG II (P < 0.05, Figure 1A and Supplemental Figure 1). We noted no differences in diastolic BP, HR, or locomotor activity in PE-infused females compared with control females (Figure 1B and Supplemental Figure 1). Similar to ANG II–treated dams, PE-treated dams had normal litter sizes (5.8 ± 0.3 pups/litter, Figure 1C) but lower P0 pup birth weights (1.26 ± 0.02 g/pup; P < 0.05, Figure 1D) relative to saline control–treated dams.
Expression of proinflammatory and microglial genes in hippocampi of young and adult mouse offspring was altered by prenatal hypertension exposure. To better understand the neurobiological changes that might drive neuropathology in hypertension-exposed offspring, we next assessed inflammatory gene expression in ANG II–exposed, PE-exposed, and control offspring brains. Hippocampi were dissected from the mice at 4 or 10 weeks of age. Male offspring from ANG II dams exhibited increased mRNA expression of proinflammatory (tumor necrosis factor [Tnf]) and microglial (cluster of differentiation molecule 11b/integrin α M [Cd11b/Itgam]) markers in the hippocampus when compared with saline controls (P < 0.05, Figure 2A and Supplemental Figure 2A). In contrast, ANG II–induced maternal hypertension had no effect on the expression of these genes in female offspring (P > 0.05, Figure 2B and Supplemental Figure 2A). Similarly, PE-induced maternal hypertension also upregulated expression of Tnf and Cd11b in hippocampi collected at 10 weeks of age in male but not female offspring (P < 0.05, Figure 2C).
 Figure 2
Figure 2Hippocampal pro-inflammation with prenatal ANG II or PE and modulation by PTX or PLX5622. Quantitative comparison of the mRNA expression of proinflammatory cytokines (Tnf) and a microglial marker (Cd11b) in the hippocampus of adult offspring (10 weeks old) from normotensive (saline-treated) dams and from dams with ANG II–induced hypertension (male offspring, A and female offspring, B) or PE-induced hypertension (offspring of both sexes, C) (n = 5–6/group). One-way ANOVA; *P < 0.05 vs. male offspring of saline-treated dams; †P < 0.05 vs. control ANG II offspring; ‡P < 0.05 vs. other 3 groups of offspring. Data are shown as the mean ± SEM.
We next evaluated the effect of pentoxifylline (PTX), a phosphodiesterase inhibitor that reduces the production of proinflammatory cytokines such as TNF in the periphery and the CNS (28–30). Two weeks of chronic PTX treatment (150 mg/kg/day in drinking water) had no effect on mRNA expression in the hippocampus of adult offspring from ANG II–treated dams (P > 0.05). In contrast, hippocampal expression of Tnf and Cd11b was significantly decreased in both male and female adult offspring of ANG II–treated dams (P < 0.05) after 1 week of treatment with the CSF 1 receptor (CSF1R) inhibitor PLX5622, which depletes microglia; in males, PLX5622 treatment restored hippocampal expression of Tnf to control levels (Figure 2, A and B).
Expression of AT1-R in adult offspring hippocampus was differentially altered in PE and ANG II hypertension models. To further differentiate the neurobiological changes in the hippocampi of ANG II–treated, PE-treated, and control offspring, we assessed the expression of 1 component of the RAS, the ANG II type 1 receptor (At1r). In hippocampi collected at 10 weeks of age, male but not female offspring of ANG II–treated dams had significantly upregulated At1r expression (P < 0.05, Supplemental Figure 3). In contrast, At1r expression in both male and female offspring of PE-treated dams was not altered. These results suggest that the maternal ANG II– and PE-induced hypertension models had different effects on the neurodevelopment and RAS programming of offspring.
Sensitivity to seizure and attendant mortality in young offspring of GH pregnancies are increased. Given the increased seizure risk in a large clinical cohort of maternal hypertension–exposed children and the increased neuroinflammation in our animal models of GH, we next examined seizure susceptibility in offspring from the ANG II model. Pilocarpine was used to induce seizures in young (4–5 weeks of age) female and male offspring, and the severity of seizures was scored on a modified Racine scale (31). There were no significant differences in Racine grades between male and female offspring from saline-treated dams following pilocarpine injections at any dose. However, both male and female offspring of ANG II–treated dams had Racine grade 3, 4, and 5 seizures following low cumulative doses (100–250 mg/kg) of pilocarpine. The Racine grades after a cumulative dose of 450–500 mg/kg of pilocarpine in male, but not female, offspring of ANG II–treated dams remained high when compared with same-sex offspring of saline-treated dams (P < 0.05, Supplemental Figure 2B). Pilocarpine injections did not cause death among these offspring.
We next used electrical stimulation (ES) as an independent method of seizure induction. Seizures and death were assessed in young offspring by applying current with stimulus intensities of 1–10 mA. Offspring from ANG II–treated dams showed a dose-dependent enhancement of Racine grades when compared with offspring from saline-treated dams (P < 0.05, Supplemental Figure 2C). Furthermore, offspring from ANG II–treated dams were more likely to die and died at a lower stimulus intensity (5 mA) than did the offspring of saline-treated dams (no deaths at 5 mA, some deaths at 10 mA; P < 0.05, Supplemental Figure 2D). There were no sex differences by either Racine grading or seizure-induced mortality, and this was true for offspring of both saline- and ANG II–treated dams.
Increased sensitivity to seizure and attendant mortality in adult mouse offspring of GH pregnancies. To complement the studies of young offspring of the ANG II model and to determine the persistence of seizure phenotypes, we next tested seizure susceptibility of adult offspring from ANG II and PE models. Like the results in young female offspring of ANG II–treated dams, pilocarpine injections induced a similar pattern of elevated Racine grades with no deaths for adult female offspring (10–12 weeks of age) of ANG II–treated dams. However, a cumulative dose of 400–500 mg/kg of pilocarpine resulted not only in significantly enhanced Racine grades, but also a 50% death rate for the male offspring of ANG II–treated dams, which was sex specific (P < 0.05, Figure 3, A and B).
 Figure 3
Figure 3Seizure induction in offspring from ANG II–treated dams. Increased sensitivity and mortality to pilocarpine-induced (A and B) or ES-induced (C and D) seizures in adult male and female offspring of dams with ANG II–induced hypertension when compared with offspring of normotensive (saline-treated) dams. Sex differences in the sensitivity and mortality induced by pilocarpine were noted (n = 7–10/group). Two-way ANOVA followed by Tukey’s test; *P < 0.05 vs. male offspring of saline-treated dams; #P < 0.05 vs. female offspring of saline-treated dams; ¥P < 0.05 vs. female offspring of ANG II–treated dams. Data are shown as the mean ± SEM.
Interestingly, we noted no sex differences in Racine grades or mortality with respect to seizures induced by ES in either adult offspring of saline-treated dams or adult offspring of ANG II–treated dams. However, Racine grades and mortality in response to ES were higher in the offspring of ANG II–treated dams (P < 0.05, Figure 3, C and D) compared with the offspring of saline-treated dams.
To confirm that ES-associated death in offspring was from seizures, we treated offspring with the antiseizure drug diazepam 30 minutes before application of ES. Diazepam prevented ES-associated death in all groups of adult offspring (Supplemental Figure 4).
To complement the ANG II model, we also performed seizure induction studies in the PE model of GH, which produces hypertension in dams with much lower systemic inflammatory effects in the dams (26). As with ANG II treatment, PE-induced GH elicited higher Racine grades and more deaths by pilocarpine administration for adult male versus female offspring (P < 0.05, Figure 4, A and B). ES also resulted in elevated Racine grades and higher mortality for both male and female offspring from the PE model (P < 0.05, Figure 4, C and D).
 Figure 4
Figure 4Seizure induction in offspring from PE–treated dams. Increased sensitivity and mortality to pilocarpine-induced (A and B) or ES-induced (C and D) seizures in adult male and female offspring of dams with PE–induced hypertension when compared with offspring of normotensive (saline-treated) dams. There were sex differences in the sensitivity and mortality induced by pilocarpine (n = 7–10/group). Two-way ANOVA followed by Tukey’s test; *P < 0.05 vs. male offspring of saline-treated dams; #P < 0.05 vs. female offspring of saline-treated dams; ¥P < 0.05 vs. female offspring of PE-treated dams. Data are shown as the mean ± SEM.
Antiinflammatory drug attenuates seizures in adult offspring of GH pregnancies. To determine the role of inflammation in GH-associated programming of offspring seizure susceptibility, we administered PTX to decrease proinflammatory cytokines in offspring from saline- or ANG II–treated dams. Two weeks of PTX administration via the drinking water did not change body weights for any group (Supplemental Figure 5, A and B). Pretreatment with PTX prior to seizure threshold assessment by the pilocarpine method had no effect in adult male offspring of saline-treated dams, but significantly attenuated seizure responses and prevented death following application of pilocarpine in adult male offspring of ANG II–treated dams (Figure 5, A and B). Similarly, PTX administered to offspring significantly reduced seizure responses to ES, as shown by Racine grades and mortality rates among male offspring of both saline- and ANG II–treated dams (P < 0.05, Figure 5, C and D). Likewise, PTX treatment also decreased seizures and mortality following pilocarpine or ES in female offspring of ANG II–treated dams (P < 0.05, Figure 6, A–C).
 Figure 5
Figure 5PTX rescue of male offspring seizure phenotypes. Pretreatment with PTX significantly attenuated seizure responses and prevented mortality after pilocarpine (A and B) or ES (C and D) treatment in adult male offspring of dams with ANG II–induced hypertension (n = 8–10/group). Two-way ANOVA followed by Tukey’s test; *P < 0.05 vs. male offspring of saline-treated dams; †P < 0.05 vs. male offspring with PTX treatment. Data are shown as the mean ± SEM.
 Figure 6
Figure 6PTX rescue of female offspring seizure phenotypes. Pretreatment with PTX significantly reduced seizure responses and mortality after pilocarpine (A) or ES (B and C) treatment in adult female offspring of both normotensive (saline-treated) dams and dams with ANG II–induced hypertension (n = 7–9/group). Two-way ANOVA followed by Tukey’s test; #P < 0.05 vs. female offspring of saline-treated dams; ‡P < 0.05 vs. PTX-treated female offspring. Data are shown as the mean ± SEM.
Pharmacologic microglia depletion attenuates seizures in adult offspring of GH pregnancies. Given the positive effects of PTX on seizure susceptibility and death in hypertension-exposed offspring, we next sought to determine the role of microglia in mediating these effects. The CSF1R inhibitor PLX5622 was administered via chow (1,200 ppm) for 1 week (32). Chow consumption and body weight were unchanged by PLX5622 administration across all groups (Supplemental Figure 5, C and D). Oral administration of PLX5622 for 7 days led to a significant depletion of microglia in the hippocampi of offspring from either saline- or ANG II–treated dams (Supplemental Figure 6, A and B). This depletion of microglia was greater in the offspring of saline-treated dams than in those of ANG II–treated dams (88.4% ± 2.3% vs 69.4% ± 3.9%, P < 0.05, Supplemental Figure 6C).
Although there was less depletion of microglia in the offspring of ANG II–treated dams, this microglial depletion was sufficient to significantly reduce seizure responses and abolish mortality following application of either pilocarpine or ES in both male (Figure 7) and female (Figure 8) offspring of ANG II–treated dams.
 Figure 7
Figure 7PLX5622 rescue of male offspring seizure phenotypes. Pretreatment with PLX5622 significantly attenuated seizure responses and abolished mortality after pilocarpine (A and B) or ES (C and D) treatment in adult male offspring of dams with ANG II–induced hypertension (n = 8–11/group). Two-way ANOVA followed by Tukey’s test; *P < 0.05 vs. male offspring of saline-treated dams; †P < 0.05 vs. male offspring with PLX 5622 treatment. Data are shown as the mean ± SEM.
 Figure 8
Figure 8PLX5622 rescue of female offspring seizure phenotypes. Pretreatment with PLX5622 significantly reduced seizure responses and mortality after pilocarpine (A) or ES (B and C) treatment in adult female offspring of dams with ANG II–induced hypertension (n = 7–9/group). Two-way ANOVA followed by Tukey’s test; #P < 0.05 vs. female offspring of saline-treated dams; ‡P < 0.05 vs. female offspring with PLX 5622 treatment. Data are shown as the mean ± SEM.













Copyright © 2026 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)