Issue published April 1, 2008 Previous issue | Next issue

- Volume 118, Issue 4
Go to section:

-
Personal perspectives
×
Abstract
For the hundredth anniversary of the American Society for Clinical Investigation, we have invited various members of the community to recount here the impact that the Society, its annual meeting, and The Journal of Clinical Investigation have had on them. Their recollections provide a view of the changes that have occurred in academic medicine in general and for the physician-scientist in particular.
Authors
Charles L. Sawyers
×Abstract
For many young physician-scientists, the American Society for Clinical Investigation spring meetings are the backdrop to their initiation into academic medicine. Membership in the ASCI is a high honor and represents one’s maturation and accomplishment in clinical research. The ASCI continues to provide this meeting forum for young investigators who aspire to emulate their idols and mentors just as I did in 1969 when I attended the spring meetings in Atlantic City for the first time.
Authors
Anthony S. Fauci
×Abstract
Congratulations to the ASCI and its membership on the first 100 years. From their first organizational meeting on the Boardwalk in Atlantic City in June 1907, our predecessors recognized the need for an organization for the clinician who had a strong interest in true experimental medicine. These nine Young Turks formed the Society, had their first meeting at the Willard Hotel in Washington, DC, in 1909, and went on to become some of the leaders of American medicine in the first half of the 20th century. The establishment of the JCI in 1924 further enhanced the high standards of the ASCI and its membership. Surely thousands of us have benefited from the foundation of the ASCI and the JCI, as have, I would submit, millions of patients as well.
Authors
William N. Kelley
×Abstract
The ASCI is notable for two unique functions — the annual meeting and The Journal of Clinical Investigation. Both have inspired us over the 37 years of our collaborative adventures in research. In this retrospective, we review highlights from our 26 joint papers in the JCI, focusing on two papers that revealed the consequences of lipid accumulation with implications for atherosclerosis and steatohepatitis.
Authors
Joseph L. Goldstein, Michael S. Brown
×Abstract
Fifty years ago, the Atlantic City meetings, held the first week in May of every year, were attended by all the elite of American academic medicine and all who wanted to join that group. Part of the magic of those meetings was that professors and neophytes took each other seriously and talked to each other.
Authors
Franklin H. Epstein
×Abstract
The annual Atlantic City meeting of the Young Turks was an exciting event — an opportunity to hear great science, to explore career opportunities, and to meet and make friends. Though the 1950s, ’60s, and ’70s, the meetings remained relatively intimate, broadly covering the best in medicine and constantly growing until the participants outgrew the Atlantic City venue to eventually spawn the numerous specialty annual meetings that we have today.
Authors
Paul A. Marks
×Abstract
In this article, I reflect on the unique value for the societies of academic internal medicine of their annual spring meetings that were held in Atlantic City for two generations prior to 1977 and consider whether lessons remain from those past experiences.
Authors
Lloyd H. Smith Jr.
×Abstract
Memories of the meetings in Atlantic City of the two major academic medical societies, the AAP and the ASCI, are enveloped by a vague and unsettling nostalgia. Dominating the scene was the Boardwalk — a site of unexpected encounters, often with long-forgotten colleagues, evoking a feeling of shared intellectual excitement and rich personal ties.
Authors
Donald W. Seldin
×Abstract
In this perspective, I trace my experiences with the ASCI, beginning in 1952, when as a medical student I attended my first meeting, until 1975, when I completed my term as president of the Society. I focus attention on the sociological aspects of the Atlantic City meetings and the critically important role these meetings played in the evolution of academic medicine during the third quarter of the 20th century.
Authors
Eugene Braunwald
×Abstract
Where once the annual meeting of the American Federation for Clinical Research, the American Society for Clinical Investigation, and the Association of American Physicians could unite the whole of clinical investigation, now stand many organizations and meetings catering to specialized fields, and the cohering effect of the Atlantic City meetings has not since been duplicated.
Authors
Arnold S. Relman
×Abstract
For many academic physician-scientists, the yearly Tri-Societies meeting of the ASCI, AAP, and AFCR during the 1960s, ’70s, and ’80s was an annual rite of spring and the focal point of the academic year. In this brief essay, I set down some miscellaneous recollections of these meetings and some thoughts about why they were of such central importance in the careers of those of my generation.
Authors
Robert J. Lefkowitz
×Abstract
The American Society for Clinical Investigation has supported the career development of physician-scientists for the past 100 years. As the ASCI looks to its next 100 years, it must be a leading force, not only for advancing the research of physician-scientists, but also for stimulating public advocacy for biomedical research in this country.
Authors
Elizabeth G. Nabel
×Abstract
I have news for the older generation who regale us with tales of presenting before the giants in Atlantic City. Pronouncements regarding the demise of such encounters are premature. The giants now convene in Chicago, but they are just as imposing.
Authors
Jonathan Epstein
×Abstract
The American Society for Clinical Investigation (ASCI) was started a century ago to foster and to address the needs of the younger physician-scientists. A hundred years later, ASCI remains one of the premier organizations for physician-scientists and one of most well-respected organizations in the medical community. I have had the opportunity and pleasure to interact with the ASCI not only as an organization through my tenure as president of the American Physician Scientists Association, but also with its members over the last four years. In my view, the same characteristics that permeate ASCI the organization also define ASCI the membership — mentorship, exemplary role models, advocacy, and leadership.
Authors
Freddy T. Nguyen








-
Review Series
×
Abstract
The most up-to-date estimates demonstrate very heterogeneous spread of HIV-1, and more than 30 million people are now living with HIV-1 infection, most of them in sub-Saharan Africa. The efficiency of transmission of HIV-1 depends primarily on the concentration of the virus in the infectious host. Although treatment with antiviral agents has proven a very effective way to improve the health and survival of infected individuals, as we discuss here, the epidemic will continue to grow unless greatly improved prevention strategies can be developed and implemented. No prophylactic vaccine is on the horizon. However, several behavioral and structural strategies have made a difference — male circumcision provides substantial protection from sexually transmitted diseases, including HIV-1, and the application of antiretroviral agents for prevention holds great promise.
Authors
Myron S. Cohen, Nick Hellmann, Jay A. Levy, Kevin DeCock, Joep Lange
×Abstract
Tuberculosis (TB) is a major threat to global health, recently exacerbated by the emergence of highly drug-resistant forms of the disease-causing pathogen and synergy with HIV/AIDS. In 2006, the Stop TB Partnership published “The global plan to stop TB: 2006–2015,” which set out a vision of halving the prevalence of and mortality caused by the disease by 2015, followed by eliminating the disease as a public health problem by 2050. This vision depends on the development of improved diagnostics, simpler treatment, and more effective vaccination. Recently, active translational research pipelines directed toward each of these goals have been established, but improved understanding of the fundamental biology of this complex disease will prove to be the key to radical advances in TB control.
Authors
Douglas B. Young, Mark D. Perkins, Ken Duncan, Clifton E. Barry III
×Abstract
There are still approximately 500 million cases of malaria and 1 million deaths from malaria each year. Yet recently, malaria incidence has been dramatically reduced in some parts of Africa by increasing deployment of anti-mosquito measures and new artemisinin-containing treatments, prompting renewed calls for global eradication. However, treatment and mosquito control currently depend on too few compounds and thus are vulnerable to the emergence of compound-resistant parasites and mosquitoes. As discussed in this Review, new drugs, vaccines, and insecticides, as well as improved surveillance methods, are research priorities. Insights into parasite biology, human immunity, and vector behavior will guide efforts to translate parasite and mosquito genome sequences into novel interventions.
Authors
Brian M. Greenwood, David A. Fidock, Dennis E. Kyle, Stefan H.I. Kappe, Pedro L. Alonso, Frank H. Collins, Patrick E. Duffy
×Abstract
Enteric infections, with or without overt diarrhea, have profound effects on intestinal absorption, nutrition, and childhood development as well as on global mortality. Oral rehydration therapy has reduced the number of deaths from dehydration caused by infection with an enteric pathogen, but it has not changed the morbidity caused by such infections. This Review focuses on the interactions between enteric pathogens and human genetic determinants that alter intestinal function and inflammation and profoundly impair human health and development. We also discuss specific implications for novel approaches to interventions that are now opened by our rapidly growing molecular understanding.
Authors
William A. Petri Jr., Mark Miller, Henry J. Binder, Myron M. Levine, Rebecca Dillingham, Richard L. Guerrant
×Abstract
Pneumonia is an illness, usually caused by infection, in which the lungs become inflamed and congested, reducing oxygen exchange and leading to cough and breathlessness. It affects individuals of all ages but occurs most frequently in children and the elderly. Among children, pneumonia is the most common cause of death worldwide. Historically, in developed countries, deaths from pneumonia have been reduced by improvements in living conditions, air quality, and nutrition. In the developing world today, many deaths from pneumonia are also preventable by immunization or access to simple, effective treatments. However, as we highlight here, there are critical gaps in our understanding of the epidemiology, etiology, and pathophysiology of pneumonia that, if filled, could accelerate the control of pneumonia and reduce early childhood mortality.
Authors
J. Anthony G. Scott, W. Abdullah Brooks, J.S. Malik Peiris, Douglas Holtzman, E. Kim Mulholland
×Abstract
Kinetoplastids are a group of flagellated protozoans that include the species Trypanosoma and Leishmania, which are human pathogens with devastating health and economic effects. The sequencing of the genomes of some of these species has highlighted their genetic relatedness and underlined differences in the diseases that they cause. As we discuss in this Review, steady progress using a combination of molecular, genetic, immunologic, and clinical approaches has substantially increased understanding of these pathogens and important aspects of the diseases that they cause. Consequently, the paths for developing additional measures to control these “neglected diseases” are becoming increasingly clear, and we believe that the opportunities for developing the drugs, diagnostics, vaccines, and other tools necessary to expand the armamentarium to combat these diseases have never been better.
Authors
Ken Stuart, Reto Brun, Simon Croft, Alan Fairlamb, Ricardo E. Gürtler, Jim McKerrow, Steve Reed, Rick Tarleton
×Abstract
Helminths are parasitic worms. They are the most common infectious agents of humans in developing countries and produce a global burden of disease that exceeds better-known conditions, including malaria and tuberculosis. As we discuss here, new insights into fundamental helminth biology are accumulating through newly completed genome projects and the nascent application of transgenesis and RNA interference technologies. At the same time, our understanding of the dynamics of the transmission of helminths and the mechanisms of the Th2-type immune responses that are induced by infection with these parasitic worms has increased markedly. Ultimately, these advances in molecular and medical helminth biology should one day translate into a new and robust pipeline of drugs, diagnostics, and vaccines for targeting parasitic worms that infect humans.
Authors
Peter J. Hotez, Paul J. Brindley, Jeffrey M. Bethony, Charles H. King, Edward J. Pearce, Julie Jacobson
×Abstract
Latest estimates indicate that nutritional deficiencies account for 3 million child deaths each year in less-developed countries. Targeted nutritional interventions could therefore save millions of lives. However, such interventions require careful optimization to maximize benefit and avoid harm. Progress toward designing effective life-saving interventions is currently hampered by some serious gaps in our understanding of nutrient metabolism in humans. In this Personal Perspective, we highlight some of these gaps and make some proposals as to how improved research methods and technologies can be brought to bear on the problems of undernourished children in the developing world.
Authors
Andrew M. Prentice, M. Eric Gershwin, Ulrich E. Schaible, Gerald T. Keusch, Cesar G. Victora, Jeffrey I. Gordon
×Abstract
Contraceptives that are readily available and acceptable are required in many poorer countries to reduce population growth and in all countries to prevent maternal morbidity and mortality arising from unintended pregnancies. Most available methods use hormonal steroids or are variations of barrier methods. Reports from several fora over the last 12 years have emphasized the number of unwanted pregnancies and resultant abortions, which indicate an unmet need for safe, acceptable, and inexpensive contraceptive methods. This unmet need can be assuaged, in part, by development of new nonhormonal contraceptive methods. This Review addresses the contribution that the “omic” revolution can make to the identification of novel contraceptive targets, as well as the progress that has been made for different target molecules under development.
Authors
R. John Aitken, Mark A. Baker, Gustavo F. Doncel, Martin M. Matzuk, Christine K. Mauck, Michael J.K. Harper








-
Commentaries
×
Abstract
In March 2006, a phase I study of the superagonistic anti-CD28 antibody TGN1412 caused a massive cytokine storm and multiorgan failure in six healthy human volunteers. Such a profound impact on the immune system was not predicted by preclinical animal studies. In a study from this issue of the JCI, Müller et al. treated rats with the superagonistic anti-CD28 antibody JJ316 and found that it rapidly induced a marked T cell lymphopenia by trapping T cells in the spleen and lymph nodes (see the related article on page 1405). This dramatic redistribution of T cells simulated the profound T cell lymphopenia observed in human recipients of TGN1412. In contrast, JJ316 treatment in the rats did not reproduce the massive cytokine storm observed following TGN1412 administration to the human volunteers. These results point to similarities as well as differences between rodents and humans in the immunological effects of superagonistic anti-CD28 antibody treatment and raise further questions about how best to design preclinical studies that can better predict the risks of novel immunotherapeutics in humans.
Authors
E. William St. Clair
×Abstract
Members of the L6 family of membrane proteins, a branch of the tetraspanin superfamily, are overexpressed in tumor cells from many types of cancers. However, direct evidence of their oncogenic activity has not been previously shown. In this issue of the JCI, Lee et al. demonstrate that overexpression of the tetraspanin superfamily member TM4SF5 in human hepatocellular carcinoma cells causes cellular phenotypic changes that resemble classical descriptions of epithelial-mesenchymal transition (EMT), with some unique aspects (see the related article beginning on page 1354). They also show that these TM4SF5-mediated effects trigger tumor formation when these cells are injected into mice. The study implicates TM4SF5, for the first time to our knowledge, in EMT oncogenic pathways of cancer progression.
Authors
Ruth J. Muschel, Annamaria Gal
×Abstract
The transcription factor homeobox B4 (HOXB4) is a promising agent capable of providing a growth advantage to genetically modified hematopoietic stem and progenitor cells (HSPCs). In this issue of the JCI, Zhang and colleagues overexpressed HOXB4 in HSPCs from large animals using retroviral vectors (see the related article beginning on page 1502). Two years after transplantation, most animals developed leukemia, a consequence of combined HOXB4 and deregulated protooncogene expression. These results highlight the risks of combining integrating vectors and growth-promoting genes for clinical applications.
Authors
Andre Larochelle, Cynthia E. Dunbar

-
Research Articles
×
Abstract
The growth of normal cells is arrested when they come in contact with each other, a process known as contact inhibition. Contact inhibition is lost during tumorigenesis, resulting in uncontrolled cell growth. Here, we investigated the role of the tetraspanin transmembrane 4 superfamily member 5 (TM4SF5) in contact inhibition and tumorigenesis. We found that TM4SF5 was overexpressed in human hepatocarcinoma tissue. TM4SF5 expression in clinical samples and in human hepatocellular carcinoma cell lines correlated with enhanced p27Kip1 expression and cytosolic stabilization as well as morphological elongation mediated by RhoA inactivation. These TM4SF5-mediated effects resulted in epithelial-mesenchymal transition (EMT) via loss of E-cadherin expression. The consequence of this was aberrant cell growth, as assessed by S-phase transition in confluent conditions, anchorage-independent growth, and tumor formation in nude mice. The TM4SF5-mediated effects were abolished by suppressing the expression of either TM4SF5 or cytosolic p27Kip1, as well as by reconstituting the expression of E-cadherin. Our observations have revealed a role for TM4SF5 in causing uncontrolled growth of human hepatocarcinoma cells through EMT.
Authors
Sin-Ae Lee, Sung-Yul Lee, Ik-Hyun Cho, Min-A Oh, Eun-Sil Kang, Yong-Bae Kim, Woo Duck Seo, Suyong Choi, Ju-Ock Nam, Mimi Tamamori-Adachi, Shigetaka Kitajima, Sang-Kyu Ye, Semi Kim, Yoon-Jin Hwang, In-San Kim, Ki Hun Park, Jung Weon Lee
×Abstract
Most patients (80%) with ovarian cancer (OvCa) present with metastatic disease. Attachment of OvCa cells to peritoneum and omentum represents the first rate-limiting step for metastatic spread. Therefore, identifying factors regulating cell attachment in the abdominal cavity is critical to the development of therapeutic agents. We show here that MMP-2 expression was upregulated in OvCa cells upon attachment to their microenvironment. Downregulation of MMP-2 mRNA or pharmacological inhibition of MMP-2 proteolytic function, in both human OvCa primary cells and cell lines, reduced attachment of OvCa cells to a 3D organotypic model of metastatic OvCa, full human omentum or peritoneum, and in vivo to mouse peritoneum and omentum. Absence of MMP-2 in the host did not alter OvCa adhesion, as determined utilizing mice harboring homozygous null mutations in either the Mmp2 or Mmp9 genes. Conversely, adhesion induced upregulation of MMP-2 mRNA in OvCa cells. MMP-2 inhibition in OvCa cells through pharmacological or antibody treatment prior to i.p. dissemination in nude mice significantly decreased tumor growth and metastasis and extended survival. MMP-2 enhanced peritoneal adhesion of OvCa cells through cleavage of ECM proteins fibronectin (FN) and vitronectin (Vn) into small fragments and increased binding of OvCa cells to these FN and Vn fragments and their receptors, α5β1 and αVβ3 integrin. These findings indicate that MMP-2 expressed by metastatic OvCa cells functionally regulates their attachment to peritoneal surfaces.
Authors
Hilary A. Kenny, Swayamjot Kaur, Lisa M. Coussens, Ernst Lengyel
×Abstract
Mobilization of endothelial progenitor cells (EPCs) from the bone marrow and their subsequent participation in neovessel formation are implicated in tumor growth and neovascularization. As the neurotransmitter dopamine (DA) modulates adult endothelial cell function, we hypothesized that DA might have a regulatory role in mobilization of EPCs from the bone marrow niche. We show that there was a significant decrease in bone marrow DA content and an increase in EPC mobilization in tumor-bearing mice associated with tumor neovascularization. DA treatment of tumor-bearing mice inhibited EPC mobilization and tumor growth through its D2 receptors, as DA treatment failed to inhibit EPC mobilization in tumor-bearing mice treated with a specific DA D2 receptor antagonist and in tumor-bearing mice lacking the D2 receptor. In addition, we found that DA, through D2 receptors, exerted its inhibitory effect on EPC mobilization through suppression of VEGFA-induced ERK1/ERK2 phosphorylation and MMP-9 synthesis. These findings reveal a new link between DA and EPC mobilization and suggest a novel use for DA and D2 agents in the treatment of cancer and other diseases involving neovessel formation.
Authors
Debanjan Chakroborty, Uttio Roy Chowdhury, Chandrani Sarkar, Rathindranath Baral, Partha Sarathi Dasgupta, Sujit Basu
×Abstract
CTLs have the potential to attack tumors, and adoptive transfer of CTLs can lead to tumor regression in mouse models and human clinical settings. However, the dynamics of tumor cell elimination during efficient T cell therapy is unknown, and it is unclear whether CTLs act directly by destroying tumor cells or indirectly by initiating the recruitment of innate immune cells that mediate tumor damage. To address these questions, we report real-time imaging of tumor cell apoptosis in vivo using intravital 2-photon microscopy and a Förster resonance energy transfer–based (FRET-based) reporter of caspase 3 activity. In a mouse model of solid tumor, we found that tumor regression after transfer of in vitro–activated CTLs occurred primarily through the direct action of CTLs on each individual tumor cell, with a minimal bystander effect. Surprisingly, the killing of 1 target cell by an individual CTL took an extended period of time, 6 hours on average, which suggested that the slow rate of killing intrinsically limits the efficiency of antitumor T cell responses. The ability to visualize when, where, and how tumor cells are killed in vivo offers new perspectives for understanding how immune effectors survey cancer cells and how local tumor microenvironments may subvert immune responses.
Authors
Béatrice Breart, Fabrice Lemaître, Susanna Celli, Philippe Bousso
×IFN-γ– and TNF-dependent bystander eradication of antigen-loss variants in established mouse cancers
Abstract
Tumors elicit antitumor immune responses, but over time they evolve and can escape immune control through various mechanisms, including the loss of the antigen to which the response is directed. The escape of antigen-loss variants (ALVs) is a major obstacle to T cell–based immunotherapy for cancer. However, cancers can be cured if both the number of CTLs and the expression of antigen are high enough to allow targeting of not only tumor cells, but also the tumor stroma. Here, we showed that IFN-γ and TNF produced by CTLs were crucial for the elimination of established mouse tumors, including ALVs. In addition, both BM- and non-BM–derived stromal cells were required to express TNF receptors and IFN-γ receptors for the elimination of ALVs. Although IFN-γ and TNF were not required by CTLs for perforin-mediated killing of antigen-expressing tumor cells, the strong inference is that tumor antigen–specific CTLs must secrete IFN-γ and TNF for destruction of tumor stroma. Therefore, bystander killing of ALVs may result from IFN-γ and TNF acting on tumor stroma.
Authors
Bin Zhang, Theodore Karrison, Donald A. Rowley, Hans Schreiber
×Abstract
Administration of the CD28 superagonistic antibody JJ316 is an efficient means to treat autoimmune diseases in rats, but the humanized antibody TGN1412 caused devastating side effects in healthy volunteers during a clinical trial. Here we show that JJ316 treatment of rats induced a dramatic redistribution of T lymphocytes from the periphery to the secondary lymphoid organs, resulting in severe T lymphopenia. Live imaging of secondary lymphoid organs revealed that JJ316 administration almost instantaneously (<2 minutes) arrested T cells in situ. This reduction in T cell motility was accompanied by profound cytoskeletal rearrangements and increased cell size. In addition, surface expression of lymphocyte function–associated antigen-1 was enhanced, endothelial differentiation sphingolipid G protein–coupled receptor 1 and L selectin levels were downregulated, and the cells lost their responsiveness to sphingosine 1–phosphate–directed migration. These proadhesive alterations were accompanied by signs of strong activation, including upregulation of CD25, CD69, CD134, and proinflammatory mediators. However, this did not lead to a cytokine storm similar to the clinical trial. While most of the early changes disappeared within 48 hours, we observed that CD4+CD25+FoxP3+ regulatory T cells experienced a second phase of activation, which resulted in massive cell enlargement, extensive polarization, and increased motility. These data suggest that CD28 superagonists elicit 2 qualitatively distinct waves of activation.
Authors
Nora Müller, Jens van den Brandt, Francesca Odoardi, Denise Tischner, Judith Herath, Alexander Flügel, Holger M. Reichardt
×Abstract
A hallmark of SLE is the production of high-titer, high-affinity, isotype-switched IgG autoantibodies directed against nucleic acid–associated antigens. Several studies have established a role for both type I IFN (IFN-I) and the activation of TLRs by nucleic acid–associated autoantigens in the pathogenesis of this disease. Here, we demonstrate that 2 IFN-I signaling molecules, IFN regulatory factor 9 (IRF9) and STAT1, were required for the production of IgG autoantibodies in the pristane-induced mouse model of SLE. In addition, levels of IgM autoantibodies were increased in pristane-treated Irf9–/– mice, suggesting that IRF9 plays a role in isotype switching in response to self antigens. Upregulation of TLR7 by IFN-α was greatly reduced in Irf9–/– and Stat1–/– B cells. Irf9–/– B cells were incapable of being activated through TLR7, and Stat1–/– B cells were impaired in activation through both TLR7 and TLR9. These data may reveal a novel role for IFN-I signaling molecules in both TLR-specific B cell responses and production of IgG autoantibodies directed against nucleic acid–associated autoantigens. Our results suggest that IFN-I is upstream of TLR signaling in the activation of autoreactive B cells in SLE.
Authors
Donna L. Thibault, Alvina D. Chu, Kareem L. Graham, Imelda Balboni, Lowen Y. Lee, Cassidy Kohlmoos, Angela Landrigan, John P. Higgins, Robert Tibshirani, Paul J. Utz
×Abstract
DNA vaccines promote an immune response by providing antigen-encoding DNA to the recipient, but the efficacy of such vaccines needs improving. Many approaches have considerable potential but currently induce relatively weak immune responses despite multiple high doses of DNA vaccine. Here, we asked whether targeting vaccine antigens to DCs would increase the immunity and protection that result from DNA vaccines. To determine this, we generated a DNA vaccine encoding a fusion protein comprised of the vaccine antigen and a single-chain Fv antibody (scFv) specific for the DC-restricted antigen-uptake receptor DEC205. Following vaccination of mice, the vaccine antigen was expressed selectively by DCs, which were required for the increased efficacy of MHC class I and MHC class II antigen presentation relative to a control scFv DNA vaccine. In addition, a DNA vaccine encoding an HIV gag p41–scFv DEC205 fusion protein induced 10-fold higher antibody levels and increased numbers of IFN-γ–producing CD4+ and CD8+ T cells. After a single i.m. injection of the DNA vaccine encoding an HIV gag p41–scFv DEC205 fusion protein, mice were protected from an airway challenge with a recombinant vaccinia virus expressing the HIV gag p41, even with 1% of the dose of nontargeted DNA vaccine. The efficacy of DNA vaccines therefore may be enhanced by inclusion of sequences such as single-chain antibodies to target the antigen to DCs.
Authors
Godwin Nchinda, Janelle Kuroiwa, Margarita Oks, Christine Trumpfheller, Chae Gyu Park, Yaoxing Huang, Drew Hannaman, Sarah J. Schlesinger, Olga Mizenina, Michel C. Nussenzweig, Klaus Überla, Ralph M. Steinman
×Abstract
Hyperkalemic periodic paralysis (HyperKPP) produces myotonia and attacks of muscle weakness triggered by rest after exercise or by K+ ingestion. We introduced a missense substitution corresponding to a human familial HyperKPP mutation (Met1592Val) into the mouse gene encoding the skeletal muscle voltage-gated Na+ channel NaV1.4. Mice heterozygous for this mutation exhibited prominent myotonia at rest and muscle fiber-type switching to a more oxidative phenotype compared with controls. Isolated mutant extensor digitorum longus muscles were abnormally sensitive to the Na+/K+ pump inhibitor ouabain and exhibited age-dependent changes, including delayed relaxation and altered generation of tetanic force. Moreover, rapid and sustained weakness of isolated mutant muscles was induced when the extracellular K+ concentration was increased from 4 mM to 10 mM, a level observed in the muscle interstitium of humans during exercise. Mutant muscle recovered from stimulation-induced fatigue more slowly than did control muscle, and the extent of recovery was decreased in the presence of high extracellular K+ levels. These findings demonstrate that expression of the Met1592Val Na+ channel in mouse muscle is sufficient to produce important features of HyperKPP, including myotonia, K+-sensitive paralysis, and susceptibility to delayed weakness during recovery from fatigue.
Authors
Lawrence J. Hayward, Joanna S. Kim, Ming-Yang Lee, Hongru Zhou, Ji W. Kim, Kumudini Misra, Mohammad Salajegheh, Fen-fen Wu, Chie Matsuda, Valerie Reid, Didier Cros, Eric P. Hoffman, Jean-Marc Renaud, Stephen C. Cannon, Robert H. Brown Jr.
×Abstract
Numerous studies have suggested that muscle atrophy is accompanied by apoptotic loss of myonuclei and therefore recovery would require replenishment by muscle stem cells. We used in vivo time-lapse microscopy to observe the loss and replenishment of myonuclei in murine muscle fibers following induced muscle atrophy. To our surprise, imaging of single fibers for up to 28 days did not support the concept of nuclear loss during atrophy. Muscles were inactivated by denervation, nerve impulse block, or mechanical unloading. Nuclei were stained in vivo either acutely by intracellular injection of fluorescent oligonucleotides or in time-lapse studies after transfection with a plasmid encoding GFP with a nuclear localization signal. We observed no loss of myonuclei in fast- or slow-twitch muscle fibers despite a greater than 50% reduction in fiber cross-sectional area. TUNEL labeling of fragmented DNA on histological sections revealed high levels of apoptotic nuclei in inactive muscles. However, when costained for laminin and dystrophin, virtually none of the TUNEL-positive nuclei could be classified as myonuclei; apoptosis was confined to stromal and satellite cells. We conclude that disuse atrophy is not a degenerative process, but is rather a change in the balance between protein synthesis and proteolysis in a permanent cell syncytium.
Authors
Jo C. Bruusgaard, Kristian Gundersen
×Abstract
Bardet-Biedl syndrome (BBS) is a heterogeneous genetic disorder characterized by many features, including obesity and cardiovascular disease. We previously developed knockout mouse models of 3 BBS genes: BBS2, BBS4, and BBS6. To dissect the mechanisms involved in the metabolic disorders associated with BBS, we assessed the development of obesity in these mouse models and found that BBS-null mice were hyperphagic, had low locomotor activity, and had elevated circulating levels of the hormone leptin. The effect of exogenous leptin on body weight and food intake was attenuated in BBS mice, which suggests that leptin resistance may contribute to hyperleptinemia. In other mouse models of obesity, leptin resistance may be selective rather than systemic; although mice became resistant to leptin’s anorectic effects, the ability to increase renal sympathetic nerve activity (SNA) was preserved. Although all 3 of the BBS mouse models were similarly resistant to leptin, the sensitivity of renal SNA to leptin was maintained in Bbs4–/– and Bbs6–/– mice, but not in Bbs2–/– mice. Consequently, Bbs4–/– and Bbs6–/– mice had higher baseline renal SNA and arterial pressure and a greater reduction in arterial pressure in response to ganglionic blockade. Furthermore, we found that BBS mice had a decreased hypothalamic expression of proopiomelanocortin, which suggests that BBS genes play an important role in maintaining leptin sensitivity in proopiomelanocortin neurons.
Authors
Kamal Rahmouni, Melissa A. Fath, Seongjin Seo, Daniel R. Thedens, Christopher J. Berry, Robert Weiss, Darryl Y. Nishimura, Val C. Sheffield
×Abstract
13-cis retinoic acid (13-cis RA; also known as isotretinoin) is the most potent agent available for treatment of acne. It is known that the drug induces apoptosis in cells cultured from human sebaceous glands, but its mechanism of action has not been determined. In this study, skin biopsies were taken from 7 patients with acne prior to and at 1 week of treatment with 13-cis RA. TUNEL staining confirmed that 13-cis RA induced apoptosis in sebaceous glands. Transcriptional profiling of patient skin and cultured human sebaceous gland cells (SEB-1 sebocytes) indicated that lipocalin 2 was among the genes most highly upregulated by 13-cis RA. Lipocalin 2 encodes neutrophil gelatinase–associated lipocalin (NGAL), which functions in innate immune defense and induces apoptosis of murine B lymphocytes. Increased immunolocalization of NGAL was noted in patients’ sebaceous glands following treatment with 13-cis RA, and recombinant NGAL induced apoptosis in SEB-1 sebocytes. Furthermore, apoptosis in response to 13-cis RA was inhibited in the presence of siRNA to lipocalin 2. These data indicate that NGAL mediates the apoptotic effect of 13-cis RA and suggest that agents that selectively induce NGAL expression in sebaceous glands might represent therapeutic alternatives to the use of 13-cis RA to treat individuals with acne.
Authors
Amanda M. Nelson, Wei Zhao, Kathryn L. Gilliland, Andrea L. Zaenglein, Wenlei Liu, Diane M. Thiboutot
×Abstract
Becoming a phenotypic male is ultimately determined by androgen-induced masculinization. Disorders of fetal masculinization, resulting in hypospadias or cryptorchidism, are common, but their cause remains unclear. Together with the adult-onset disorders low sperm count and testicular cancer, they can constitute a testicular dysgenesis syndrome (TDS). Although masculinization is well studied, no unifying concept explains normal male reproductive development and its abnormalities, including TDS. We exposed rat fetuses to either anti-androgens or androgens and showed that masculinization of all reproductive tract tissues was programmed by androgen action during a common fetal programming window. This preceded morphological differentiation, when androgen action was, surprisingly, unnecessary. Only within the programming window did blocking androgen action induce hypospadias and cryptorchidism and altered penile length in male rats, all of which correlated with anogenital distance (AGD). Androgen-driven masculinization of females was also confined to the same programming window. This work has identified in rats a common programming window in which androgen action is essential for normal reproductive tract masculinization and has highlighted that measuring AGD in neonatal humans could provide a noninvasive method to predict neonatal and adult reproductive disorders. Based on the timings in rats, we believe the programming window in humans is likely to be 8–14 weeks of gestation.
Authors
Michelle Welsh, Philippa T.K. Saunders, Mark Fisken, Hayley M. Scott, Gary R. Hutchison, Lee B. Smith, Richard M. Sharpe
×Abstract
Priapism, abnormally prolonged penile erection in the absence of sexual excitation, is associated with ischemia-mediated erectile tissue damage and subsequent erectile dysfunction. It is common among males with sickle cell disease (SCD), and SCD transgenic mice are an accepted model of the disorder. Current strategies to manage priapism suffer from a poor fundamental understanding of the molecular mechanisms underlying the disorder. Here we report that mice lacking adenosine deaminase (ADA), an enzyme necessary for the breakdown of adenosine, displayed unexpected priapic activity. ADA enzyme therapy successfully corrected the priapic activity both in vivo and in vitro, suggesting that it was dependent on elevated adenosine levels. Further genetic and pharmacologic evidence demonstrated that A2B adenosine receptor–mediated (A2BR-mediated) cAMP and cGMP induction was required for elevated adenosine–induced prolonged penile erection. Finally, priapic activity in SCD transgenic mice was also caused by elevated adenosine levels and A2BR activation. Thus, we have shown that excessive adenosine accumulation in the penis contributes to priapism through increased A2BR signaling in both Ada–/– and SCD transgenic mice. These findings provide insight regarding the molecular basis of priapism and suggest that strategies to either reduce adenosine or block A2BR activation may prove beneficial in the treatment of this disorder.
Authors
Tiejuan Mi, Shahrzad Abbasi, Hong Zhang, Karen Uray, Janci L. Chunn, Ling Wei Xia, Jose G. Molina, Norman W. Weisbrodt, Rodney E. Kellems, Michael R. Blackburn, Yang Xia
×Abstract
Retroviral vector–mediated HSC gene therapy has been used to treat individuals with a number of life-threatening diseases. However, some patients with SCID-X1 developed retroviral vector–mediated leukemia after treatment. The selective growth advantage of gene-modified cells in patients with SCID-X1 suggests that the transgene may have played a role in leukemogenesis. Here we report that 2 of 2 dogs and 1 of 2 macaques developed myeloid leukemia approximately 2 years after being transplanted with cells that overexpressed homeobox B4 (HOXB4) and cells transduced with a control gammaretroviral vector that did not express HOXB4. The leukemic cells had dysregulated expression of oncogenes, a block in myeloid differentiation, and overexpression of HOXB4. HOXB4 knockdown restored differentiation in leukemic cells, suggesting involvement of HOXB4. In contrast, leukemia did not arise from the cells carrying the control gammaretroviral vector. In addition, leukemia did not arise in 5 animals with high-level marking and polyclonal long-term repopulation following transplantation with cells transduced with an identical gammaretrovirus vector backbone expressing methylguanine methyltransferase. These findings, combined with the absence of leukemia in many other large animals transplanted with cells transduced with gammaretroviral vectors expressing genes other than HOXB4, show that HOXB4 overexpression poses a significant risk of leukemogenesis. Our data thus suggest the continued need for caution in genetic manipulation of repopulating cells, particularly when the transgene might impart an intrinsic growth advantage.
Authors
Xiao-Bing Zhang, Brian C. Beard, Grant D. Trobridge, Brent L. Wood, George E. Sale, Reeteka Sud, R. Keith Humphries, Hans-Peter Kiem
×Abstract
Deficiencies in the SBDS gene result in Shwachman-Diamond syndrome (SDS), an inherited bone marrow failure syndrome associated with leukemia predisposition. SBDS encodes a highly conserved protein previously implicated in ribosome biogenesis. Using human primary bone marrow stromal cells (BMSCs), lymphoblasts, and skin fibroblasts, we show that SBDS stabilized the mitotic spindle to prevent genomic instability. SBDS colocalized with the mitotic spindle in control primary BMSCs, lymphoblasts, and skin fibroblasts and bound to purified microtubules. Recombinant SBDS protein stabilized microtubules in vitro. We observed that primary BMSCs and lymphoblasts from SDS patients exhibited an increased incidence of abnormal mitoses. Similarly, depletion of SBDS by siRNA in human skin fibroblasts resulted in increased mitotic abnormalities and aneuploidy that accumulated over time. Treatment of primary BMSCs and lymphoblasts from SDS patients with nocodazole, a microtubule destabilizing agent, led to increased mitotic arrest and apoptosis, consistent with spindle destabilization. Conversely, SDS patient cells were resistant to taxol, a microtubule stabilizing agent. These findings suggest that spindle instability in SDS contributes to bone marrow failure and leukemogenesis.
Authors
Karyn M. Austin, Mohan L. Gupta Jr., Scott A. Coats, Asmin Tulpule, Gustavo Mostoslavsky, Alejandro B. Balazs, Richard C. Mulligan, George Daley, David Pellman, Akiko Shimamura
×Abstract
Dominant mutations in the gene encoding the mRNA splicing factor PRPF31 cause retinitis pigmentosa, a hereditary form of retinal degeneration. Most of these mutations are characterized by DNA changes that lead to premature termination codons. We investigated 6 different PRPF31 mutations, represented by single-base substitutions or microdeletions, in cell lines derived from 9 patients with dominant retinitis pigmentosa. Five of these mutations lead to premature termination codons, and 1 leads to the skipping of exon 2. Allele-specific measurement of PRPF31 transcripts revealed a strong reduction in the expression of mutant alleles. As a consequence, total PRPF31 protein abundance was decreased, and no truncated proteins were detected. Subnuclear localization of the full-length PRPF31 that was present remained unaffected. Blocking nonsense-mediated mRNA decay significantly restored the amount of mutant PRPF31 mRNA but did not restore the synthesis of mutant proteins, even in conjunction with inhibitors of protein degradation pathways. Our results indicate that most PRPF31 mutations ultimately result in null alleles through the activation of surveillance mechanisms that inactivate mutant mRNA and, possibly, proteins. Furthermore, these data provide compelling evidence that the pathogenic effect of PRPF31 mutations is likely due to haploinsufficiency rather than to gain of function.
Authors
Thomas Rio Frio, Nicholas M. Wade, Adriana Ransijn, Eliot L. Berson, Jacques S. Beckmann, Carlo Rivolta
×Abstract
Axonal degeneration is an important determinant of progressive neurological disability in multiple sclerosis (MS). Thus, therapeutic approaches promoting neuroprotection could aid the treatment of progressive MS. Here, we used what we believe is a novel water-soluble fullerene derivative (ABS-75) attached to an NMDA receptor antagonist, which combines antioxidant and anti-excitotoxic properties, to block axonal damage and reduce disease progression in a chronic progressive EAE model. Fullerene ABS-75 treatment initiated after disease onset reduced the clinical progression of chronic EAE in NOD mice immunized with myelin-oligodendrocyte glycoprotein (MOG). Reduced disease progression in ABS-75–treated mice was associated with reduced axonal loss and demyelination in the spinal cord. Fullerene ABS-75 halted oxidative injury, CD11b+ infiltration, and CCL2 expression in the spinal cord of mice without interfering with antigen-specific T cell responses. In vitro, fullerene ABS-75 protected neurons from oxidative and glutamate-induced injury and restored glutamine synthetase and glutamate transporter expression in astrocytes under inflammatory insult. Glutamine synthetase expression was also increased in the white matter of fullerene ABS-75–treated animals. Our data demonstrate the neuroprotective effect of treatment with a fullerene compound combined with a NMDA receptor antagonist, which may be useful in the treatment of progressive MS and other neurodegenerative diseases.
Authors
Alexandre S. Basso, Dan Frenkel, Francisco J. Quintana, Frederico A. Costa-Pinto, Sanja Petrovic-Stojkovic, Lindsay Puckett, Alon Monsonego, Amnon Bar-Shir, Yoni Engel, Michael Gozin, Howard L. Weiner
×Abstract
The essential contribution of the antidepressant-sensitive serotonin (5-HT) transporter SERT (which is encoded by the SLC6A4 gene) to platelet 5-HT stores suggests an important role of this transporter in platelet function. Here, using SERT-deficient mice, we have established a role for constitutive SERT expression in efficient ADP- and thrombin-triggered platelet aggregation. Additionally, using pharmacological blockers of SERT and the vesicular monoamine transporter (VMAT), we have identified a role for ongoing 5-HT release and SERT activity in efficient human platelet aggregation. We have also demonstrated that fibrinogen, an activator of integrin αIIbβ3, enhances SERT activity in human platelets and that integrin αIIbβ3 interacts directly with the C terminus of SERT. Consistent with these findings, knockout mice lacking integrin β3 displayed diminished platelet SERT activity. Conversely, HEK293 cells engineered to express human SERT and an activated form of integrin β3 exhibited enhanced SERT function that coincided with elevated SERT surface expression. Our results support an unsuspected role of αIIbβ3/SERT associations as well as αIIbβ3 activation in control of SERT activity in vivo that may have broad implications for hyperserotonemia, cardiovascular disorders, and autism.
Authors
Ana Marin D. Carneiro, Edwin H. Cook, Dennis L. Murphy, Randy D. Blakely
×Abstract
Nontyphoidal strains of Salmonella (NTS) are a common cause of bacteremia among African children. Cell-mediated immune responses control intracellular infection, but they do not protect against extracellular growth of NTS in the blood. We investigated whether antibody protects against NTS bacteremia in Malawian children, because we found this condition mainly occurs before 2 years of age, with relative sparing of infants younger than 4 months old. Sera from all healthy Malawian children tested aged more than 16 months contained anti-Salmonella antibody and successfully killed NTS. Killing was mediated by complement membrane attack complex and not augmented in the presence of blood leukocytes. Sera from most healthy children less than 16 months old lacked NTS-specific antibody, and sera lacking antibody did not kill NTS despite normal complement function. Addition of Salmonella-specific antibody, but not mannose-binding lectin, enabled NTS killing. All NTS strains tested had long-chain lipopolysaccharide and the rck gene, features that resist direct complement-mediated killing. Disruption of lipopolysaccharide biosynthesis enabled killing of NTS by serum lacking Salmonella-specific antibody. We conclude that Salmonella-specific antibody that overcomes the complement resistance of NTS develops by 2 years of life in Malawian children. This finding and the age-incidence of NTS bacteremia suggest that antibody protects against NTS bacteremia and support the development of vaccines against NTS that induce protective antibody.
Authors
Calman A. MacLennan, Esther N. Gondwe, Chisomo L. Msefula, Robert A. Kingsley, Nicholas R. Thomson, Sarah A. White, Margaret Goodall, Derek J. Pickard, Stephen M. Graham, Gordon Dougan, C. Anthony Hart, Malcolm E. Molyneux, Mark T. Drayson
×Abstract
The hormone estradiol affects the auditory system both by itself and by its interaction with neuroprotective factors. In this study, we examined the role of estrogen receptors (ERs) in response to auditory trauma. We found a ligand-dependent protective role for ERβ in the auditory system by investigating mice deficient in ERα (ERKO mice), ERβ (BERKO mice), and aromatase (ARKO mice). Basal auditory brainstem response (ABR) thresholds were similar in all animals. An acoustic trauma causing a temporary hearing loss raised ABR thresholds in male and female BERKO and ARKO mice compared with WT and ERKO mice. The ERα-selective agonist, propyl(1H) pyrazole-1,3,5-triyl-trisphenol (PPT), partially protected ARKO mice from trauma, while the ERβ-selective agonist, 2,3-bis (4-hydroxyphenyl)-propionitrile (DPN), protected WT and ARKO mice. Immunohistochemistry and western blotting confirmed the expression of ERβ in cochlea of WT males and females. Levels of brain-derived neurotrophic factor (BDNF), a neuroprotective peptide that can be induced by estrogen, was lower in BERKO and ARKO mice compared with WT. DPN treatment increased BDNF expression in ARKO mice. These data indicate ERβ-mediated neuroprotection involving BDNF in the auditory system of males and females.
Authors
Inna Meltser, Yeasmin Tahera, Evan Simpson, Malou Hultcrantz, Konstantina Charitidi, Jan-Åke Gustafsson, Barbara Canlon
×Abstract
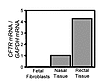
Progress toward understanding the pathogenesis of cystic fibrosis (CF) and developing effective therapies has been hampered by lack of a relevant animal model. CF mice fail to develop the lung and pancreatic disease that cause most of the morbidity and mortality in patients with CF. Pigs may be better animals than mice in which to model human genetic diseases because their anatomy, biochemistry, physiology, size, and genetics are more similar to those of humans. However, to date, gene-targeted mammalian models of human genetic disease have not been reported for any species other than mice. Here we describe the first steps toward the generation of a pig model of CF. We used recombinant adeno-associated virus (rAAV) vectors to deliver genetic constructs targeting the CF transmembrane conductance receptor (CFTR) gene to pig fetal fibroblasts. We generated cells with the CFTR gene either disrupted or containing the most common CF-associated mutation (ΔF508). These cells were used as nuclear donors for somatic cell nuclear transfer to porcine oocytes. We thereby generated heterozygote male piglets with each mutation. These pigs should be of value in producing new models of CF. In addition, because gene-modified mice often fail to replicate human diseases, this approach could be used to generate models of other human genetic diseases in species other than mice.
Authors
Christopher S. Rogers, Yanhong Hao, Tatiana Rokhlina, Melissa Samuel, David A. Stoltz, Yuhong Li, Elena Petroff, Daniel W. Vermeer, Amanda C. Kabel, Ziying Yan, Lee Spate, David Wax, Clifton N. Murphy, August Rieke, Kristin Whitworth, Michael L. Linville, Scott W. Korte, John F. Engelhardt, Michael J. Welsh, Randall S. Prather
×Abstract
Somatic cell gene targeting combined with nuclear transfer cloning presents tremendous potential for the creation of new, large-animal models of human diseases. Mouse disease models often fail to reproduce human phenotypes, underscoring the need for the generation and study of alternative disease models. Mice deficient for CFTR have been poor models for cystic fibrosis (CF), lacking many aspects of human CF lung disease. In this study, we describe the production of a CFTR gene–deficient model in the domestic ferret using recombinant adeno-associated virus–mediated gene targeting in fibroblasts, followed by nuclear transfer cloning. As part of this approach, we developed a somatic cell rejuvenation protocol using serial nuclear transfer to produce live CFTR-deficient clones from senescent gene-targeted fibroblasts. We transferred 472 reconstructed embryos into 11 recipient jills and obtained 8 healthy male ferret clones heterozygous for a disruption in exon 10 of the CFTR gene. To our knowledge, this study represents the first description of genetically engineered ferrets and describes an approach that may be of substantial utility in modeling not only CF, but also other genetic diseases.
Authors
Xingshen Sun, Ziying Yan, Yaling Yi, Ziyi Li, Diana Lei, Christopher S. Rogers, Juan Chen, Yulong Zhang, Michael J. Welsh, Gregory H. Leno, John F. Engelhardt

























Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)