Issue published July 3, 2006 Previous issue | Next issue

- Volume 116, Issue 7
Go to section:

-
Editorial
×
Abstract
Scientists are usually thought to be beyond reproach, but with the recent spate of high-profile ethical transgressions by scientists, the public’s trust in science and scientists is deteriorating. The numerous cases of scientific misconduct that have crossed my desk in the last year leave me disenchanted, disappointed, and disillusioned.
Authors
Ushma S. Neill

-
Science in Medicine
×
Abstract
Parkinson disease (PD) is a relatively common disorder of the nervous system that afflicts patients later in life with tremor, slowness of movement, gait instability, and rigidity. Treatment of these cardinal features of the disease is a success story of modern science and medicine, as a great deal of disability can be alleviated through the pharmacological correction of brain dopamine deficiency. Unfortunately these therapies only provide temporary, though significant, relief from early symptoms and do not halt disease progression. In addition, pathological changes outside of the motor system leading to cognitive, autonomic, and psychiatric symptoms are not sufficiently treated by current therapies. Much as the discovery of dopamine deficiency led to powerful treatments for motor symptoms, recent discoveries concerning the role of specific genes in PD pathology will lead to the next revolution in disease therapy. Understanding why and how susceptible cells in motor and nonmotor regions of the brain die in PD is the first step toward preventing this cell death and curing or slowing the disease. In this review we discuss recent discoveries in the fields of diagnosis and treatment of PD and focus on how a better understanding of disease mechanisms gained through the study of monogenetic forms of PD has provided novel therapeutic targets.
Authors
Joseph M. Savitt, Valina L. Dawson, Ted M. Dawson

-
Review Series Introduction
×
Abstract
It is now well accepted that diabetes mellitus is one of the main threats to human health in the twenty-first century. The total number of people with diabetes worldwide was estimated at between 151 million and 171 million in 2000 and is projected to increase to 221 million in 2010 and to 366 million in 2030. Needless to say, the increase in the number of people with diabetes will be accompanied by an increase in the number of those with diabetic complications such as nephropathy, retinopathy, neuropathy, and atherosclerosis. The global mortality attributable to diabetes in the year 2000 was estimated at 2.9 million deaths, a number that will also increase. Given that type 2 diabetes accounts for more than 90% of cases of diabetes worldwide, it is important that we understand the pathogenesis of this condition and develop new approaches to its prevention and treatment.
Authors
Masato Kasuga

-
Review Series
×
Abstract
Insulin has pleiotropic biological effects in virtually all tissues. However, the relevance of insulin signaling in peripheral tissues has been studied far more extensively than its role in the brain. An evolving body of evidence indicates that in the brain, insulin is involved in multiple regulatory mechanisms including neuronal survival, learning, and memory, as well as in regulation of energy homeostasis and reproductive endocrinology. Here we review insulin’s role as a central homeostatic signal with regard to energy and glucose homeostasis and discuss the mechanisms by which insulin communicates information about the body’s energy status to the brain. Particular emphasis is placed on the controversial current debate about the similarities and differences between hypothalamic insulin and leptin signaling at the molecular level.
Authors
Leona Plum, Bengt F. Belgardt, Jens C. Brüning
×Abstract
Recent data underscore the importance of intertissue communication in the maintenance of normal glucose homeostasis. Important signals are conveyed by hormones, cytokines, and fuel substrates and are sensed through a variety of cellular mechanisms. The ability of tissues to sense and adapt to changes in metabolic status and fuel availability is altered in insulin-resistant states including type 2 diabetes. Here we review the roles of glucose and its metabolites as signaling molecules and the diverse physiologic mechanisms for glucose sensing.
Authors
Mark A. Herman, Barbara B. Kahn
×Abstract
AMP-activated protein kinase (AMPK) is an energy sensor that regulates cellular metabolism. When activated by a deficit in nutrient status, AMPK stimulates glucose uptake and lipid oxidation to produce energy, while turning off energy-consuming processes including glucose and lipid production to restore energy balance. AMPK controls whole-body glucose homeostasis by regulating metabolism in multiple peripheral tissues, such as skeletal muscle, liver, adipose tissues, and pancreatic β cells — key tissues in the pathogenesis of type 2 diabetes. By responding to diverse hormonal signals including leptin and adiponectin, AMPK serves as an intertissue signal integrator among peripheral tissues, as well as the hypothalamus, in the control of whole-body energy balance.
Authors
Yun Chau Long, Juleen R. Zierath
×Abstract
Adiponectin is an adipokine that is specifically and abundantly expressed in adipose tissue and directly sensitizes the body to insulin. Hypoadiponectinemia, caused by interactions of genetic factors such as SNPs in the Adiponectin gene and environmental factors causing obesity, appears to play an important causal role in insulin resistance, type 2 diabetes, and the metabolic syndrome, which are linked to obesity. The adiponectin receptors, AdipoR1 and AdipoR2, which mediate the antidiabetic metabolic actions of adiponectin, have been cloned and are downregulated in obesity-linked insulin resistance. Upregulation of adiponectin is a partial cause of the insulin-sensitizing and antidiabetic actions of thiazolidinediones. Therefore, adiponectin and adiponectin receptors represent potential versatile therapeutic targets to combat obesity-linked diseases characterized by insulin resistance. This Review describes the pathophysiology of adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome.
Authors
Takashi Kadowaki, Toshimasa Yamauchi, Naoto Kubota, Kazuo Hara, Kohjiro Ueki, Kazuyuki Tobe
×Abstract
Over a hundred years ago, high doses of salicylates were shown to lower glucose levels in diabetic patients. This should have been an important clue to link inflammation to the pathogenesis of type 2 diabetes (T2D), but the antihyperglycemic and antiinflammatory effects of salicylates were not connected to the pathogenesis of insulin resistance until recently. Together with the discovery of an important role for tissue macrophages, these new findings are helping to reshape thinking about how obesity increases the risk for developing T2D and the metabolic syndrome. The evolving concept of insulin resistance and T2D as having immunological components and an improving picture of how inflammation modulates metabolism provide new opportunities for using antiinflammatory strategies to correct the metabolic consequences of excess adiposity.
Authors
Steven E. Shoelson, Jongsoon Lee, Allison B. Goldfine
×Abstract
The major focus of this Review is on the mechanisms of islet β cell failure in the pathogenesis of obesity-associated type 2 diabetes (T2D). As this demise occurs within the context of β cell compensation for insulin resistance, consideration is also given to the mechanisms involved in the compensation process, including mechanisms for expansion of β cell mass and for enhanced β cell performance. The importance of genetic, intrauterine, and environmental factors in the determination of “susceptible” islets and overall risk for T2D is reviewed. The likely mechanisms of β cell failure are discussed within the two broad categories: those with initiation and those with progression roles.
Authors
Marc Prentki, Christopher J. Nolan
×Abstract
Considerable evidence supports the association between insulin resistance and vascular disease, and this has led to wide acceptance of the clustering of hyperlipidemia, glucose intolerance, hypertension, and obesity as a clinical entity, the metabolic syndrome. While insulin resistance, by promoting dyslipidemia and other metabolic abnormalities, is part of the proatherogenic milieu, it is possible that insulin resistance itself in the vascular wall does not promote atherosclerosis. Recent findings suggest that insulin resistance and atherosclerosis could represent independent and ultimately maladaptive responses to the disruption of cellular homeostasis caused by the excess delivery of fuel.
Authors
Clay F. Semenkovich






-
Commentaries
×
Abstract
Predicting the chances of recovery of consciousness and communication in patients who survive their coma but transit in a vegetative state or minimally conscious state (MCS) remains a major challenge for their medical caregivers. Very few studies have examined the slow neuronal changes underlying functional recovery of consciousness from severe chronic brain damage. A case study in this issue of the JCI reports an extraordinary recovery of functional verbal communication and motor function in a patient who remained in MCS for 19 years (see the related article beginning on page 2005). Diffusion tensor MRI showed increased fractional anisotropy (assumed to reflect myelinated fiber density) in posteromedial cortices, encompassing cuneus and precuneus. These same areas showed increased glucose metabolism as studied by PET scanning, likely reflecting the neuronal regrowth paralleling the patient’s clinical recovery. This case shows that old dogmas need to be oppugned, as recovery with meaningful reduction in disability continued in this case for nearly 2 decades after extremely severe traumatic brain injury.
Authors
Steven Laureys, Mélanie Boly, Pierre Maquet
×Abstract
Mutations in genes encoding desmosomal proteins have been identified as the major cause of arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVC), in which the right ventricle is “replaced” by fibrofatty tissue, resulting in lethal arrhythmias. In this issue of the JCI, Garcia-Gras et al. demonstrate that cardiac-specific loss of the desmosomal protein desmoplakin is sufficient to cause nuclear translocation of plakoglobin, upregulation of adipogenic genes in vitro, and a shift from a cardiomyocyte to an adipocyte cell fate in vivo (see the related article beginning on page 2012). This evidence for potential Wnt/β-catenin signaling defects sets the scene for a comprehensive exploration of the contributions of this pathway to the pathophysiology of ARVC, not only through perturbation of cardiac patterning and development, but also through effects on myocardial differentiation and physiology.
Authors
Calum A. MacRae, Walter Birchmeier, Ludwig Thierfelder
×Abstract
Congenital hydrocephalus affects 0.1–0.3% of live births, with a high mortality rate (~50%) in the absence of surgical intervention. Although the insertion of shunts alleviates the symptoms of the majority of congenital cases, the molecular basis of hydrocephalus and the mechanisms of cerebrospinal fluid (CSF) circulation remain largely unknown. Two important players are the subcommissural organ/Reissner’s fiber (SCO/RF) complex and the ventricular ependymal (vel) cells that together facilitate the flow of the CSF through the narrow canals of the ventricular system. In this issue of the JCI, Lang et al. demonstrate that overexpression of the pituitary adenylate cyclase–activating polypeptide (PACAP) type I (PAC1) receptor gene results in abnormal development of the SCO and vel cells, leading to congenital hydrocephalus (see the related article beginning on page 1924). The ligand for the PAC1 receptor is the neuropeptide PACAP, which uncovers what the authors believe to be a novel role for this signaling cascade in the regulation of CSF circulation.
Authors
David J. Picketts
×Abstract
Liver X receptors (LXRs) broadly limit cholesterol accumulation by regulating expression of genes involved in cholesterol efflux and storage. In this issue of the JCI, Cummins et al. report that LXRα is involved in similar regulation in the adrenal cortex, but it also substantially modulates glucocorticoid synthesis (see the related article beginning on page 1902). LXRα deletion in mice increases the availability of adrenal cholesterol for steroid synthesis by decreasing the expression of cholesterol efflux transporters. Glucocorticoid synthesis requires intramitochondrial cholesterol transport mediated by the steroidogenic acute regulatory protein (StAR). Surprisingly, LXR deletion and stimulation by an agonist each increase glucocorticoid synthesis. This parallels increased expression of StAR and several other steroidogenic genes.
Authors
Colin R. Jefcoate
×Abstract
Adenosine, long known as a regulator of cardiovascular function, has recently been identified as a significant paracrine inhibitor of inflammation that acts primarily by activation of A2A adenosine receptors (A2AARs) on lymphoid or myeloid cells. In this issue of the JCI, Yang et al. describe a proinflammatory phenotype resulting from deletion of the gene encoding the A2B adenosine receptor (A2BAR) in the mouse, suggesting that activation of the A2BAR can also have antiinflammatory effects (see the related article beginning on page 1913). Nevertheless, the role of the A2BAR remains enigmatic since its activation can either stimulate or inhibit the release of proinflammatory cytokines in different cells and tissues.
Authors
Joel Linden
×Abstract
Several clinical trials of bone marrow stem cell therapy for myocardial infarction are ongoing, but the mechanistic basis for any potential therapeutic effect is currently unclear. A growing body of evidence suggests that the potential improvement in cardiac function is largely independent of cardiac muscle regeneration. A study by Fazel et al. in this issue of the JCI provides evidence that bone marrow–derived c-kit+ cells can lead to an improvement in cardiac function in mutant hypomorphic c-kit mice that is independent of transdifferentiation into either cardiac muscle or endothelial cells, but rather is associated with the release of angiogenic cytokines and associated neovascularization in the infarct border zone (see the related article beginning on page 1865). These findings suggest the potential therapeutic effect of specific paracrine pathways for angiogenesis in improving cardiac function in the injured heart.
Authors
Kenneth R. Chien
×Abstract
Gene therapy is an attractive approach for the treatment of hemophilia, as continuous expression of donated clotting factor VIII (FVIII) DNA would ensure clotting factor replacement at constant circulating levels rather than at the peaks and troughs that characterize the current protein infusion therapeutic approach. In this issue of the JCI, Shi et al. describe an interesting variant of a gene transfer approach for hemophilia (see the related article beginning on page 1974). They show that targeted expression of FVIII in megakaryocytes, with storage in the α-granules of platelets, has the advantage of delivering clotting factors directly to the site of an injury, where platelets accumulate in large numbers and undergo activation accompanied by release of granule contents. Earlier clinical experience with gene transfer into hematopoietic cells highlighted the potential safety risks of this approach, but an F8 transgene may represent a lower risk than transgenes for growth factors or their receptors.
Authors
Katherine A. High







-
Research Articles
×
Abstract
Cholangiocellular carcinoma (CC), the second most common primary liver cancer, is associated with a poor prognosis. It has been shown that CCs harbor alterations of a number of tumor-suppressor genes and oncogenes, yet key regulators for tumorigenesis remain unknown. Here we have generated a mouse model that develops CC with high penetrance using liver-specific targeted disruption of tumor suppressors SMAD4 and PTEN. In the absence of SMAD4 and PTEN, hyperplastic foci emerge exclusively from bile ducts of mutant mice at 2 months of age and continue to grow, leading to tumor formation in all animals at 4–7 months of age. We show that CC formation follows a multistep progression of histopathological changes that are associated with significant alterations, including increased levels of phosphorylated AKT, FOXO1, GSK-3β, mTOR, and ERK and increased nuclear levels of cyclin D1. We further demonstrate that SMAD4 and PTEN regulate each other through a novel feedback mechanism to maintain an expression balance and synergistically repress CC formation. Finally, our analysis of human CC detected PTEN inactivation in a majority of p-AKT–positive CCs, while about half also lost SMAD4 expression. These findings elucidate the relationship between SMAD4 and PTEN and extend our understanding of CC formation.
Authors
Xiaoling Xu, Shogo Kobayashi, Wenhui Qiao, Cuiling Li, Cuiying Xiao, Svetlana Radaeva, Bangyan Stiles, Rui-Hong Wang, Nobuya Ohara, Tadashi Yoshino, Derek LeRoith, Michael S. Torbenson, Gregory J. Gores, Hong Wu, Bin Gao, Chu-Xia Deng
×Abstract
Class IIa histone deacetylases (HDACs) regulate a variety of cellular processes, including cardiac growth, bone development, and specification of skeletal muscle fiber type. Multiple serine/threonine kinases control the subcellular localization of these HDACs by phosphorylation of common serine residues, but whether certain class IIa HDACs respond selectively to specific kinases has not been determined. Here we show that calcium/calmodulin-dependent kinase II (CaMKII) signals specifically to HDAC4 by binding to a unique docking site that is absent in other class IIa HDACs. Phosphorylation of HDAC4 by CaMKII promotes nuclear export and prevents nuclear import of HDAC4, with consequent derepression of HDAC target genes. In cardiomyocytes, CaMKII phosphorylation of HDAC4 results in hypertrophic growth, which can be blocked by a signal-resistant HDAC4 mutant. These findings reveal a central role for HDAC4 in CaMKII signaling pathways and have implications for the control of gene expression by calcium signaling in a variety of cell types.
Authors
Johannes Backs, Kunhua Song, Svetlana Bezprozvannaya, Shurong Chang, Eric N. Olson
×Abstract
Clinical trials of bone marrow stem/progenitor cell therapy after myocardial infarction (MI) have shown promising results, but the mechanism of benefit is unclear. We examined the nature of endogenous myocardial repair that is dependent on the function of the c-kit receptor, which is expressed on bone marrow stem/progenitor cells and on recently identified cardiac stem cells. MI increased the number of c-kit+ cells in the heart. These cells were traced back to a bone marrow origin, using genetic tagging in bone marrow chimeric mice. The recruited c-kit+ cells established a proangiogenic milieu in the infarct border zone by increasing VEGF and by reversing the cardiac ratio of angiopoietin-1 to angiopoietin-2. These oscillations potentiated endothelial mitogenesis and were associated with the establishment of an extensive myofibroblast-rich repair tissue. Mutations in the c-kit receptor interfered with the mobilization of the cells to the heart, prevented angiogenesis, diminished myofibroblast-rich repair tissue formation, and led to precipitous cardiac failure and death. Replacement of the mutant bone marrow with wild-type cells rescued the cardiomyopathic phenotype. We conclude that, consistent with their documented role in tumorigenesis, bone marrow c-kit+ cells act as key regulators of the angiogenic switch in infarcted myocardium, thereby driving efficient cardiac repair.
Authors
Shafie Fazel, Massimo Cimini, Liwen Chen, Shuhong Li, Denis Angoulvant, Paul Fedak, Subodh Verma, Richard D. Weisel, Armand Keating, Ren-Ke Li
×Abstract
We found that sterile wounding of human skin induced epidermal expression of the antimicrobial (poly)peptides human β-defensin–3, neutrophil gelatinase–associated lipocalin, and secretory leukocyte protease inhibitor through activation of the epidermal growth factor receptor. After skin wounding, the receptor was activated by heparin-binding epidermal growth factor that was released by a metalloprotease-dependent mechanism. Activation of the epidermal growth factor receptor generated antimicrobial concentrations of human β-defensin–3 and increased the activity of organotypic epidermal cultures against Staphylococcus aureus. These data demonstrate that sterile wounding initiates an innate immune response that increases resistance to overt infection and microbial colonization.
Authors
Ole E. Sørensen, Dharma R. Thapa, K. Markus Roupé, Erika V. Valore, Ulf Sjöbring, Alice A. Roberts, Artur Schmidtchen, Tomas Ganz
×Abstract
Leptin and insulin have been identified as fuel sensors acting in part through their hypothalamic receptors to inhibit food intake and stimulate energy expenditure. As their intracellular signaling converges at the PI3K pathway, we directly addressed the role of phosphatidylinositol3,4,5-trisphosphate–mediated (PIP3-mediated) signals in hypothalamic proopiomelanocortin (POMC) neurons by inactivating the gene for the PIP3 phosphatase Pten specifically in this cell type. Here we show that POMC-specific disruption of Pten resulted in hyperphagia and sexually dimorphic diet-sensitive obesity. Although leptin potently stimulated Stat3 phosphorylation in POMC neurons of POMC cell–restricted Pten knockout (PPKO) mice, it failed to significantly inhibit food intake in vivo. POMC neurons of PPKO mice showed a marked hyperpolarization and a reduction in basal firing rate due to increased ATP-sensitive potassium (KATP) channel activity. Leptin was not able to elicit electrical activity in PPKO POMC neurons, but application of the PI3K inhibitor LY294002 and the KATP blocker tolbutamide restored electrical activity and leptin-evoked firing of POMC neurons in these mice. Moreover, icv administration of tolbutamide abolished hyperphagia in PPKO mice. These data indicate that PIP3-mediated signals are critical regulators of the melanocortin system via modulation of KATP channels.
Authors
Leona Plum, Xiaosong Ma, Brigitte Hampel, Nina Balthasar, Roberto Coppari, Heike Münzberg, Marya Shanabrough, Denis Burdakov, Eva Rother, Ruth Janoschek, Jens Alber, Bengt F. Belgardt, Linda Koch, Jost Seibler, Frieder Schwenk, Csaba Fekete, Akira Suzuki, Tak W. Mak, Wilhelm Krone, Tamas L. Horvath, Frances M. Ashcroft, Jens C. Brüning
×Abstract
Cholesterol is the obligate precursor to adrenal steroids but is cytotoxic at high concentrations. Here, we show the role of the liver X receptors (LXRα and LXRβ) in preventing accumulation of free cholesterol in mouse adrenal glands by controlling expression of genes involved in all aspects of cholesterol utilization, including the steroidogenic acute regulatory protein, StAR, a novel LXR target. Under chronic dietary stress, adrenal glands from Lxrαβ–/– mice accumulated free cholesterol. In contrast, wild-type animals maintained cholesterol homeostasis through basal expression of genes involved in cholesterol efflux and storage (ABC transporter A1 [ABCA1], apoE, SREBP-1c) while preventing steroidogenic gene (StAR) expression. Upon treatment with an LXR agonist that mimics activation by oxysterols, expression of these target genes was increased. Basally, Lxrαβ–/– mice exhibited a marked decrease in ABCA1 and a derepression of StAR expression, causing a net decrease in cholesterol efflux and an increase in steroidogenesis. These changes occurred under conditions that prevented the acute stress response and resulted in a phenotype more specific to the loss of LXRα, including hypercorticosteronemia, cholesterol ester accumulation, and adrenomegaly. These results imply LXRα provides a safety valve to limit free cholesterol levels as a basal protective mechanism in the adrenal gland, where cholesterol is under constant flux.
Authors
Carolyn L. Cummins, David H. Volle, Yuan Zhang, Jeffrey G. McDonald, Benoît Sion, Anne-Marie Lefrançois-Martinez, Françoise Caira, Georges Veyssière, David J. Mangelsdorf, Jean-Marc A. Lobaccaro
×Abstract
Adenosine has been described as playing a role in the control of inflammation, but it has not been certain which of its receptors mediate this effect. Here, we generated an A2B adenosine receptor–knockout/reporter gene–knock-in (A2BAR-knockout/reporter gene–knock-in) mouse model and showed receptor gene expression in the vasculature and macrophages, the ablation of which causes low-grade inflammation compared with age-, sex-, and strain-matched control mice. Augmentation of proinflammatory cytokines, such as TNF-α, and a consequent downregulation of IκB-α are the underlying mechanisms for an observed upregulation of adhesion molecules in the vasculature of these A2BAR-null mice. Intriguingly, leukocyte adhesion to the vasculature is significantly increased in the A2BAR-knockout mice. Exposure to an endotoxin results in augmented proinflammatory cytokine levels in A2BAR-null mice compared with control mice. Bone marrow transplantations indicated that bone marrow (and to a lesser extent vascular) A2BARs regulate these processes. Hence, we identify the A2BAR as a new critical regulator of inflammation and vascular adhesion primarily via signals from hematopoietic cells to the vasculature, focusing attention on the receptor as a therapeutic target.
Authors
Dan Yang, Ying Zhang, Hao G. Nguyen, Milka Koupenova, Anil K. Chauhan, Maria Makitalo, Matthew R. Jones, Cynthia St. Hilaire, David C. Seldin, Paul Toselli, Edward Lamperti, Barbara M. Schreiber, Haralambos Gavras, Denisa D. Wagner, Katya Ravid
×Abstract
Hydrocephalus is a common and potentially devastating birth defect affecting the CNS, and its relationship with G protein–coupled receptors (GPCRs) is unknown. We have expressed 2, 4, or 6 copies of a GPCR — the human PAC1 receptor with a 130-kb transgene in the mouse nervous system in a pattern closely resembling that of the endogenous gene. Consistent with PAC1 actions, PKA and PKC activity were elevated in the brains of Tg mice. Remarkably, Tg mice developed dose-dependent hydrocephalus-like characteristics, including enlarged third and lateral ventricles and reduced cerebral cortex, corpus callosum, and subcommissural organ (SCO). Neuronal proliferation and apoptosis were implicated in hydrocephalus, and we observed significantly reduced neuronal proliferation and massively increased neuronal apoptosis in the developing cortex and SCO of Tg embryos, while neurite outgrowth and neuronal migration in vitro remain uncompromised. Ventricular ependymal cilia are crucial for directing cerebrospinal fluid flow, and ependyma of Tg mice exhibited disrupted cilia with increased phospho-CREB immunoreactivity. These data demonstrate that altered neuronal proliferation/apoptosis and disrupted ependymal cilia are the main factors contributing to hydrocephalus in PAC1-overexpressing mice. This is the first report to our knowledge demonstrating that misregulation of GPCRs can be involved in hydrocephalus-related neurodevelopmental disorders.
Authors
Bing Lang, Bing Song, Wendy Davidson, Alastair MacKenzie, Norman Smith, Colin D. McCaig, Anthony J. Harmar, Sanbing Shen
×Abstract
CTL-associated antigen 4 (CTLA4) blockade releases inhibitory controls on T cell activation and proliferation, inducing antitumor immunity in both preclinical and early clinical trials. We examined the mechanisms of action of anti-CTLA4 and a GM-CSF–transduced tumor cell vaccine (Gvax) and their impact on the balance of effector T cells (Teffs) and Tregs in an in vivo model of B16/BL6 melanoma. Tumor challenge increased the frequency of Tregs in lymph nodes, and untreated tumors became infiltrated by CD4+Foxp3– and CD4+Foxp3+ T cells but few CD8+ T cells. Anti-CTLA4 did not deplete Tregs or permanently impair their function but acted in a cell-intrinsic manner on both Tregs and Teffs, allowing them to expand, most likely in response to self antigen. While Gvax primed the tumor-reactive Teff compartment, inducing activation, tumor infiltration, and a delay in tumor growth, the combination with CTLA4 blockade induced greater infiltration and a striking change in the intratumor balance of Tregs and Teffs that directly correlated with tumor rejection. The data suggest that Tregs control both CD4+ and CD8+ T cell activity within the tumor, highlight the importance of the intratumor ratio of effectors to regulators, and demonstrate inversion of the ratio and correlation with tumor rejection during Gvax/anti-CTLA4 immunotherapy.
Authors
Sergio A. Quezada, Karl S. Peggs, Michael A. Curran, James P. Allison
×Abstract
Bacterial vectors may offer many advantages over other antigen delivery systems for cancer vaccines. We engineered a Salmonella typhimuriumvaccine strain to deliver the NY-ESO-1 tumor antigen (S. typhimurium–NY-ESO-1) through a type III protein secretion system. The S. typhimurium–NY-ESO-1 construct elicited NY-ESO-1–specific CD8+ and CD4+ T cells from peripheral blood lymphocytes ofcancer patients in vitro. Oral administration of S. typhimurium–NY-ESO-1 to mice resulted in the regression of established NY-ESO-1–expressing tumors. Intratumoral inoculation of S. typhimurium–NY-ESO-1 to NY-ESO-1–negative tumors resulted in delivery of antigen in vivo and led to tumor regression in the presence of preexisting NY-ESO-1–specific CD8+ T cells. Specific T cell responses against at least 2 unrelated tumor antigens not contained in the vaccine were observed, demonstrating epitope spreading. We propose that antigen delivery through the S. typhimuriumtype III secretion system is a promising novel strategy for cancer vaccine development.
Authors
Hiroyoshi Nishikawa, Eiichi Sato, Gabriel Briones, Li-Mei Chen, Mitsutoshi Matsuo, Yasuhiro Nagata, Gerd Ritter, Elke Jäger, Hideki Nomura, Shigeto Kondo, Isao Tawara, Takuma Kato, Hiroshi Shiku, Lloyd J. Old, Jorge E. Galán, Sacha Gnjatic
×Abstract
Tumor-associated fibroblasts are key regulators of tumorigenesis. In contrast to tumor cells, which are genetically unstable and mutate frequently, the presence of genetically more stable fibroblasts in the tumor-stromal compartment makes them an optimal target for cancer immunotherapy. These cells are also the primary source of collagen type I, which contributes to decreased chemotherapeutic drug uptake in tumors and plays a significant role in regulating tumor sensitivity to a variety of chemotherapies. To specifically kill tumor-associated fibroblasts, we constructed an oral DNA vaccine targeting fibroblast activation protein (FAP), which is specifically overexpressed by fibroblasts in the tumor stroma. Through CD8+ T cell–mediated killing of tumor-associated fibroblasts, our vaccine successfully suppressed primary tumor cell growth and metastasis of multidrug-resistant murine colon and breast carcinoma. Furthermore, tumor tissue of FAP-vaccinated mice revealed markedly decreased collagen type I expression and up to 70% greater uptake of chemotherapeutic drugs. Most importantly, pFap-vaccinated mice treated with chemotherapy showed a 3-fold prolongation in lifespan and marked suppression of tumor growth, with 50% of the animals completely rejecting a tumor cell challenge. This strategy opens a new venue for the combination of immuno- and chemotherapies.
Authors
Markus Loeffler, Jörg A. Krüger, Andreas G. Niethammer, Ralph A. Reisfeld
×Abstract
To develop an animal model of Kaposi sarcoma–associated herpesvirus (KSHV) infection uniquely suited to evaluate longitudinal patterns of viral gene expression, cell tropism, and immune responses, we injected NOD/SCID mice intravenously with purified virus and measured latent and lytic viral transcripts in distal organs over the subsequent 4 months. We observed sequential escalation of first latent and then lytic KSHV gene expression coupled with electron micrographic evidence of virion production within the murine spleen. Using novel technology that integrates flow cytometry with immunofluorescence microscopy, we found that the virus establishes infection in murine B cells, macrophages, NK cells, and, to a lesser extent, dendritic cells. To investigate the potential for human KSHV–specific immune responses within this immunocompromised host, we implanted NOD/SCID mice with functional human hematopoietic tissue grafts (NOD/SCID-hu mice) and observed that a subset of animals produced human KSHV–specific antibodies. Furthermore, treatment of these chimeric mice with ganciclovir at the time of inoculation led to prolonged but reversible suppression of KSHV DNA and RNA levels, suggesting that KSHV can establish latent infection in vivo despite ongoing suppression of lytic replication.
Authors
Christopher H. Parsons, Laura A. Adang, Jon Overdevest, Christine M. O’Connor, J. Robert Taylor, David Camerini, Dean H. Kedes
×Abstract
Inhibitory immune response to exogenously infused factor VIII (FVIII) is a major complication in the treatment of hemophilia A. Generation of such inhibitors has the potential to disrupt gene therapy for hemophilia A. We explore what we believe to be a novel approach to overcome this shortcoming. Human B-domain–deleted FVIII (hBDDFVIII) was expressed under the control of the platelet-specific αIIb promoter in platelets of hemophilic (FVIIInull) mice to create 2bF8trans mice. The FVIII transgene product was stored in platelets and released at the site of platelet activation. In spite of the lack of FVIII in the plasma of 2bF8trans mice, the bleeding phenotype of FVIIInull mice was corrected. More importantly, the bleeding phenotype was corrected in the presence of high inhibitory antibody titers introduced into the mice by infusion or by spleen cell transfer from recombinant hBDDFVIII–immunized mice. Our results demonstrate that this approach to the targeted expression of FVIII in platelets has the potential to correct hemophilia A, even in the presence of inhibitory immune responses to infused FVIII.
Authors
Qizhen Shi, David A. Wilcox, Scot A. Fahs, Hartmut Weiler, Clive W. Wells, Brian C. Cooley, Drashti Desai, Patricia A. Morateck, Jack Gorski, Robert R. Montgomery
×Abstract
Many homeostatic processes, including appetite and food intake, are controlled by neuroendocrine circuits involving the CNS. The CNS also directly regulates adipocyte metabolism, as we have shown here by examining central action of the orexigenic hormone ghrelin. Chronic central ghrelin infusion resulted in increases in the glucose utilization rate of white and brown adipose tissue without affecting skeletal muscle. In white adipocytes, mRNA expression of various fat storage–promoting enzymes such as lipoprotein lipase, acetyl-CoA carboxylase α, fatty acid synthase, and stearoyl-CoA desaturase–1 was markedly increased, while that of the rate-limiting step in fat oxidation, carnitine palmitoyl transferase–1α, was decreased. In brown adipocytes, central ghrelin infusion resulted in lowered expression of the thermogenesis-related mitochondrial uncoupling proteins 1 and 3. These ghrelin effects were dose dependent, occurred independently from ghrelin-induced hyperphagia, and seemed to be mediated by the sympathetic nervous system. Additionally, the expression of some fat storage enzymes was decreased in ghrelin-deficient mice, which led us to conclude that central ghrelin is of physiological relevance in the control of cell metabolism in adipose tissue. These results unravel the existence of what we believe to be a new CNS-based neuroendocrine circuit regulating metabolic homeostasis of adipose tissue.
Authors
Claudia Theander-Carrillo, Petra Wiedmer, Philippe Cettour-Rose, Ruben Nogueiras, Diego Perez-Tilve, Paul Pfluger, Tamara R. Castaneda, Patrick Muzzin, Annette Schürmann, Ildiko Szanto, Matthias H. Tschöp, Françoise Rohner-Jeanrenaud
×Abstract
Resistance to chemotherapy presents a serious challenge in the successful treatment of various cancers and is mainly responsible for mortality associated with disseminated cancers. Here we show that expression of HtrA1, which is frequently downregulated in ovarian cancer, influences tumor response to chemotherapy by modulating chemotherapy-induced cytotoxicity. Downregulation of HtrA1 attenuated cisplatin- and paclitaxel-induced cytotoxicity, while forced expression of HtrA1 enhanced cisplatin- and paclitaxel-induced cytotoxicity. HtrA1 expression was upregulated by both cisplatin and paclitaxel treatment. This upregulation resulted in limited autoproteolysis and activation of HtrA1. Active HtrA1 induces cell death in a serine protease–dependent manner. The potential role of HtrA1 as a predictive factor of clinical response to chemotherapy was assessed in both ovarian and gastric cancer patients receiving cisplatin-based regimens. Patients with ovarian or gastric tumors expressing higher levels of HtrA1 showed a higher response rate compared with those with lower levels of HtrA1 expression. These findings uncover what we believe to be a novel pathway by which serine protease HtrA1 mediates paclitaxel- and cisplatin-induced cytotoxicity and suggest that loss of HtrA1 in ovarian and gastric cancers may contribute to in vivo chemoresistance.
Authors
Jeremy Chien, Giovanni Aletti, Alfonso Baldi, Vincenzo Catalano, Pietro Muretto, Gary L. Keeney, Kimberly R. Kalli, Julie Staub, Michael Ehrmann, William A. Cliby, Yean Kit Lee, Keith C. Bible, Lynn C. Hartmann, Scott H. Kaufmann, Viji Shridhar
×Abstract
We used diffusion tensor imaging (DTI) to study 2 patients with traumatic brain injury. The first patient recovered reliable expressive language after 19 years in a minimally conscious state (MCS); the second had remained in MCS for 6 years. Comparison of white matter integrity in the patients and 20 normal subjects using histograms of apparent diffusion constants and diffusion anisotropy identified widespread altered diffusivity and decreased anisotropy in the damaged white matter. These findings remained unchanged over an 18-month interval between 2 studies in the first patient. In addition, in this patient, we identified large, bilateral regions of posterior white matter with significantly increased anisotropy that reduced over 18 months. In contrast, notable increases in anisotropy within the midline cerebellar white matter in the second study correlated with marked clinical improvements in motor functions. This finding was further correlated with an increase in resting metabolism measured by PET in this subregion. Aberrant white matter structures were evident in the second patient’s DTI images but were not clinically correlated. We propose that axonal regrowth may underlie these findings and provide a biological mechanism for late recovery. Our results are discussed in the context of recent experimental studies that support this inference.
Authors
Henning U. Voss, Aziz M. Uluç, Jonathan P. Dyke, Richard Watts, Erik J. Kobylarz, Bruce D. McCandliss, Linda A. Heier, Bradley J. Beattie, Klaus A. Hamacher, Shankar Vallabhajosula, Stanley J. Goldsmith, Douglas Ballon, Joseph T. Giacino, Nicholas D. Schiff
×Abstract
Arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVC) is a genetic disease caused by mutations in desmosomal proteins. The phenotypic hallmark of ARVC is fibroadipocytic replacement of cardiac myocytes, which is a unique phenotype with a yet-to-be-defined molecular mechanism. We established atrial myocyte cell lines expressing siRNA against desmoplakin (DP), responsible for human ARVC. We show suppression of DP expression leads to nuclear localization of the desmosomal protein plakoglobin and a 2-fold reduction in canonical Wnt/β-catenin signaling through Tcf/Lef1 transcription factors. The ensuing phenotype is increased expression of adipogenic and fibrogenic genes and accumulation of fat droplets. We further show that cardiac-restricted deletion of Dsp, encoding DP, impairs cardiac morphogenesis and leads to high embryonic lethality in the homozygous state. Heterozygous DP-deficient mice exhibited excess adipocytes and fibrosis in the myocardium, increased myocyte apoptosis, cardiac dysfunction, and ventricular arrhythmias, thus recapitulating the phenotype of human ARVC. We believe our results provide for a novel molecular mechanism for the pathogenesis of ARVC and establish cardiac-restricted DP-deficient mice as a model for human ARVC. These findings could provide for the opportunity to identify new diagnostic markers and therapeutic targets in patients with ARVC.
Authors
Eduardo Garcia-Gras, Raffaella Lombardi, Michael J. Giocondo, James T. Willerson, Michael D. Schneider, Dirar S. Khoury, Ali J. Marian
×Abstract
CD4+CD25+ Tregs regulate immunity, but little is known about their own regulation. We now report that the human 60-kDa heat shock protein (HSP60) acts as a costimulator of human Tregs, both CD4+CD25int and CD4+CD25hi. Treatment of Tregs with HSP60, or its peptide p277, before anti-CD3 activation significantly enhanced the ability of relatively low concentrations of the Tregs to downregulate CD4+CD25– or CD8+ target T cells, detected as inhibition of target T cell proliferation and IFN-γ and TNF-α secretion. The enhancing effects of HSP60 costimulation on Tregs involved innate signaling via TLR2, led to activation of PKC, PI3K, and p38, and were further enhanced by inhibition of ERK. HSP60-treated Tregs suppressed target T cells both by cell-to-cell contact and by secretion of TGF-β and IL-10. In addition, the expression of ERK, NF-κB, and T-bet by downregulated target T cells was inhibited. Thus, HSP60, a self-molecule, can downregulate adaptive immune responses by upregulating Tregs innately through TLR2 signaling.
Authors
Alexandra Zanin-Zhorov, Liora Cahalon, Guy Tal, Raanan Margalit, Ofer Lider, Irun R. Cohen
×Abstract
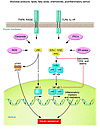
Neutrophil NADPH oxidase plays a key role in host defense and in inflammation by releasing large amounts of superoxide and other ROSs. Proinflammatory cytokines such as GM-CSF and TNF-α prime ROS production by neutrophils through unknown mechanisms. Here we used peptide sequencing by tandem mass spectrometry to show that GM-CSF and TNF-α induce phosphorylation of Ser345 on p47phox, a cytosolic component of NADPH oxidase, in human neutrophils. As Ser345 is located in the MAPK consensus sequence, we tested the effects of MAPK inhibitors. Inhibitors of the ERK1/2 pathway abrogated GM-CSF–induced phosphorylation of Ser345, while p38 MAPK inhibitor abrogated TNF-α–induced phosphorylation of Ser345. Transfection of HL-60 cells with a mutated p47phox (S345A) inhibited GM-CSF– and TNF-α–induced priming of ROS production. This event was also inhibited in neutrophils by a cell-permeable peptide containing a TAT-p47phox-Ser345 sequence. Furthermore, ROS generation, p47phox-Ser345 phosphorylation, and ERK1/2 and p38 MAPK phosphorylation were increased in synovial neutrophils from rheumatoid arthritis (RA) patients, and TAT-Ser345 peptide inhibited ROS production by these primed neutrophils. This study therefore identifies convergent MAPK pathways on Ser345 that are involved in GM-CSF– and TNF-α–induced priming of neutrophils and are activated in RA. Inhibition of the point of convergence of these pathways might serve as a novel antiinflammatory strategy.
Authors
Pham My-Chan Dang, Allan Stensballe, Tarek Boussetta, Houssam Raad, Cedric Dewas, Yolande Kroviarski, Gilles Hayem, Ole N. Jensen, Marie-Anne Gougerot-Pocidalo, Jamel El-Benna
×Abstract
The IL-21 receptor (IL-21R) shows significant homology with the IL-4R, and CD4+ Th2 cells are an important source of IL-21. Here we examined whether the IL-21R regulates the development of Th2 responses in vivo. To do this, we infected IL-21R–/– mice with the Th2-inducing pathogens Schistosoma mansoni and Nippostrongylus brasiliensis and examined the influence of IL-21R deficiency on the development of Th2-dependent pathology. We showed that granulomatous inflammation and liver fibrosis were significantly reduced in S. mansoni–infected IL-21R–/– mice and in IL-21R+/+ mice treated with soluble IL-21R–Fc (sIL-21R–Fc). The impaired granulomatous response was also associated with a marked reduction in Th2 cytokine expression and function, as evidenced by the attenuated IL-4, IL-13, AMCase, Ym1, and FIZZ1 (also referred to as RELMα) responses in the tissues. A similarly impaired Th2 response was observed following N. brasiliensis infection. In vitro, IL-21 significantly augmented IL-4Rα and IL-13Rα1 expression in macrophages, resulting in increased FIZZ1 mRNA and arginase-1 activity following stimulation with IL-4 and IL-13. As such, these data identify the IL-21R as an important amplifier of alternative macrophage activation. Collectively, these results illustrate an essential function for the IL-21R in the development of pathogen-induced Th2 responses, which may have relevance in therapies for both inflammatory and chronic fibrotic diseases.
Authors
John Pesce, Mallika Kaviratne, Thirumalai R. Ramalingam, Robert W. Thompson, Joseph F. Urban, Allen W. Cheever, Deborah A. Young, Mary Collins, Michael J. Grusby, Thomas A. Wynn























Copyright © 2025 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)