Advertisement
Genetic counseling throughout the life cycle
Leslie J. Ciarleglio,1 Robin L. Bennett,2 Jennifer Williamson,3 Jessica B. Mandell,4 and Joan H. Marks5
1Yale University School of Medicine, New Haven, Connecticut, USA2 Medical Genetics Clinic, University of Washington, Seattle, Washington, USA3 Gertrude H. Sergievsky Center, Columbia University College of Physicians and Surgeons, New York, New York, USA4 King Laboratory, Departments of Medicine and Genome Sciences, University of Washington, Seattle, Washington, USA5 Human Genetics Program, Sarah Lawrence College, Bronxville, New York, USA
Address correspondence to: Robin L. Bennett, University of Washington, Medical Genetics, Box 357720, Seattle, Washington 98195-7720, USA. Phone: (206) 616-2135; Fax: (206) 616-2414; E-mail: robinb@u.washington.edu.
Find articles by Ciarleglio, L. in: JCI | PubMed | Google Scholar
1Yale University School of Medicine, New Haven, Connecticut, USA2 Medical Genetics Clinic, University of Washington, Seattle, Washington, USA3 Gertrude H. Sergievsky Center, Columbia University College of Physicians and Surgeons, New York, New York, USA4 King Laboratory, Departments of Medicine and Genome Sciences, University of Washington, Seattle, Washington, USA5 Human Genetics Program, Sarah Lawrence College, Bronxville, New York, USA
Address correspondence to: Robin L. Bennett, University of Washington, Medical Genetics, Box 357720, Seattle, Washington 98195-7720, USA. Phone: (206) 616-2135; Fax: (206) 616-2414; E-mail: robinb@u.washington.edu.
Find articles by Bennett, R. in: JCI | PubMed | Google Scholar
1Yale University School of Medicine, New Haven, Connecticut, USA2 Medical Genetics Clinic, University of Washington, Seattle, Washington, USA3 Gertrude H. Sergievsky Center, Columbia University College of Physicians and Surgeons, New York, New York, USA4 King Laboratory, Departments of Medicine and Genome Sciences, University of Washington, Seattle, Washington, USA5 Human Genetics Program, Sarah Lawrence College, Bronxville, New York, USA
Address correspondence to: Robin L. Bennett, University of Washington, Medical Genetics, Box 357720, Seattle, Washington 98195-7720, USA. Phone: (206) 616-2135; Fax: (206) 616-2414; E-mail: robinb@u.washington.edu.
Find articles by Williamson, J. in: JCI | PubMed | Google Scholar
1Yale University School of Medicine, New Haven, Connecticut, USA2 Medical Genetics Clinic, University of Washington, Seattle, Washington, USA3 Gertrude H. Sergievsky Center, Columbia University College of Physicians and Surgeons, New York, New York, USA4 King Laboratory, Departments of Medicine and Genome Sciences, University of Washington, Seattle, Washington, USA5 Human Genetics Program, Sarah Lawrence College, Bronxville, New York, USA
Address correspondence to: Robin L. Bennett, University of Washington, Medical Genetics, Box 357720, Seattle, Washington 98195-7720, USA. Phone: (206) 616-2135; Fax: (206) 616-2414; E-mail: robinb@u.washington.edu.
Find articles by Mandell, J. in: JCI | PubMed | Google Scholar
1Yale University School of Medicine, New Haven, Connecticut, USA2 Medical Genetics Clinic, University of Washington, Seattle, Washington, USA3 Gertrude H. Sergievsky Center, Columbia University College of Physicians and Surgeons, New York, New York, USA4 King Laboratory, Departments of Medicine and Genome Sciences, University of Washington, Seattle, Washington, USA5 Human Genetics Program, Sarah Lawrence College, Bronxville, New York, USA
Address correspondence to: Robin L. Bennett, University of Washington, Medical Genetics, Box 357720, Seattle, Washington 98195-7720, USA. Phone: (206) 616-2135; Fax: (206) 616-2414; E-mail: robinb@u.washington.edu.
Find articles by Marks, J. in: JCI | PubMed | Google Scholar
Published November 1, 2003 - More info
J Clin Invest. 2003;112(9):1280–1286. https://doi.org/10.1172/JCI20170.
© 2003 The American Society for Clinical Investigation
Genetic counseling is an expanding field in the era of genomic medicine. This unique medical specialty provides clinical health care, education, and emotional support to individuals and families facing genetic and inherited diseases. Genetic counselors provide services to patients across the lifespan by assessing family and environmental history to determine disease risk; assisting in genetic testing, diagnosis, and disease prevention and management; and offering psychosocial and ethical guidance to help patients make informed, autonomous health care and reproductive decisions (Figure 1). The purpose of this article is to provide an authoritative review of genetic counseling, its role in the front lines of genetic health, and its impact on medical research, education, and patient care.
 Figure 1
Figure 1Genetic counseling is a unique medical specialty that provides clinical health care for patients across the lifespan facing genetic and inherited diseases.
-
Prenatal genetic counseling
For pregnancy-related issues, there are two general types of genetic counseling sessions — those where parents or prospective parents are concerned about potential risks and outcomes of pregnancy, and those where patients are dealing with a specific fetal diagnosis during pregnancy (17). Despite the vastly different nature of these sessions, their courses are similar. All clients come to genetic counseling with some level of anxiety, even in the most routine of circumstances. Fear of the investigation of one’s own basic genetic make-up is common, particularly in the prenatal setting. The risk of a problem, or the diagnosis of a specific anomaly during pregnancy, is often a prospective parent’s biggest fear. This anxiety can be overwhelming and may significantly impact a patient’s self-image. Such issues need to be explored, acknowledged, and accounted for throughout the counseling session.
Prior to the initial counseling appointment for an already identified fetal anomaly like heart disease, the counselor will ideally have had the opportunity to contact the family to prepare them for the discussion and to begin to develop a rapport. At the initial visit, parents are often asked to recount how the diagnosis was made and what information they have obtained thus far. This allows the family members to “tell their story” in their own words. Such recounting of family history helps the counselor assess their understanding of the diagnosis, their informational resources, and their coping strategies. It also fosters trust between the counselor and the clients.
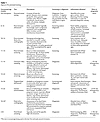
Immediately after the diagnosis of a fetal cardiac anomaly, most families are not in the frame of mind to construct a detailed three-generation pedigree. The counselor obtains an abbreviated version by asking about family and obstetrical history, use of medications, and illnesses or other exposures in pregnancy (18). The specific fetal cardiac diagnosis, including implications, severity, and natural history, is discussed in detail, as well as options for further testing and for prenatal management. All of these are influenced by gestational age at the time of the diagnosis (Table 1).
After considering the available options, the counselor and the clients determine a management plan. In addition to providing appropriate medical and emotional support, a management plan gives families some sense of control where many feel like victims in a situation beyond their control (13). Further testing, consultations with pediatric cardiologists, nurses, and surgeons, and tours of delivery suites and special-care nurseries may be planned (5, 6). Additionally, lay literature and guidance about appropriate Internet sites and other resources are critical for families and reinforce issues covered in the session (19).
-
Genetic predisposition to adult-onset disease
The identification of susceptibility genes for common adult-onset genetic diseases is moving the field of genetic counseling in a new and challenging direction. Common diseases such as diabetes, certain cancers, and adult-onset neurodegenerative diseases have an identified genetic component or have been linked to specific chromosomal regions through family linkage and association studies (Table 2). Genetic counseling for these diseases is difficult when the disease is linked to susceptibility genes, which are known to confer an increased risk of disease, rather than deterministic genes, which are predictive of disease onset. With the identification of genes associated with Alzheimer disease, there is considerable interest in the clinical application of this genetic information through genetic counseling and testing.

Article tools
- Download citation information
- Send a comment
- Terms of use
- Standard abbreviations
- Need help? Email the journal
Metrics
Go to
- Top
- Abstract
- Prenatal diagnosis: congenital heart disease
- Prenatal genetic counseling
- Treating CHD
- Exploring postdiagnosis options
- Pediatric genetic disease and newborn screening
- The challenges of screening and counseling
- Genetic predisposition to adult-onset disease
- Alzheimer disease
- To test or not to test
- Benefits of predisposition genetic testing
- Footnotes
- References
- Version history



Copyright © 2024 American Society for Clinical Investigation
ISSN: 0021-9738 (print), 1558-8238 (online)